
Abstract
Background:
Managing OFF episodes in patients with Parkinson’s disease becomes increasingly challenging over time, making it critical to tailor treatment to each patient’s needs and characteristics for effective care.
Objectives:
Study CTH-301 assessed the long-term safety/tolerability and efficacy of sublingual apomorphine (SL-APO) for the on-demand treatment of OFF episodes.
Design:
The findings from four post hoc analyses of Study CTH-301, conducted to understand factors influencing SL-APO retention and safety/tolerability, with a particular focus on oropharyngeal treatment-emergent adverse events (TEAEs) are reported.
Methods:
The first analysis evaluated baseline variables differing between patients who completed the study and those who discontinued due to either lack of efficacy or adverse events to help define patients more likely to benefit from SL-APO therapy: The second and third analyses compared safety/tolerability between the subgroups of patients who were or were not receiving dopamine agonist (DA) treatment, and in those aged <70 or ⩾70 years at baseline, respectively. The fourth analysis examined oropharyngeal TEAEs.
Results:
Patients in a younger age group, those experiencing morning akinesia or delayed ON, and those taking lower dose/fewer intakes of levodopa and concomitant DAs were more likely to benefit from SL-APO therapy. Patients taking concomitant DAs reported lower rates of DA-related TEAEs and a higher mean SL-APO optimal dose. Specific analyses in patients aged ⩾70 years indicated that this age group reported similar rates of TEAEs and a similar profile of the most common TEAEs compared with the group aged <70 years. A lower total daily dose of SL-APO was associated with a reduced risk of developing oropharyngeal TEAEs. Such events were mostly mild or moderate, occurring within the first months after SL-APO initiation, and generally resolved, with worsening being rare.
Conclusion:
These analyses provided insights into retention and safety/tolerability of SL-APO, helping clinicians and patients make informed treatment decisions.
Keywords
Introduction
Apomorphine sublingual film (SL-APO) was shown to be generally well-tolerated and efficacious as an on-demand treatment for OFF episodes in patients with Parkinson’s disease (PD) in a phase III, 12-week, double-blind, placebo-controlled trial (Study CTH-300; ClinicalTrials.gov, NCT02469090). 1 Subsequently, Study CTH-301 (a phase III, multicentre, open-label study) confirmed the efficacy and safety/tolerability of SL-APO as an on-demand treatment for OFF episodes in patients with PD over the long term (⩾3 years). 2 Based on this clinical trial evidence, SL-APO is indicated in adult patients with PD for the intermittent treatment of OFF episodes that are not sufficiently controlled by oral PD medication. 3 After receiving the first dose of SL-APO in the clinic, patients are able to identify their optimal dose when self-administering SL-APO at home. 4
Treatment-emergent adverse events (TEAEs) were a significant cause of study discontinuation among patients treated with SL-APO. In Study CTH-300, mild-to-moderate oropharyngeal side effects were the most frequently reported TEAEs, affecting 31% (17/54) of patients who received SL-APO, compared with 7% (4/55) of those who received placebo. 1 Oropharyngeal side effects also accounted for most of the discontinuations over the long term in Study CTH-301: of the 426 patients included in the long-term safety phase, 19% discontinued due to oropharyngeal side effects, with the most frequent specific reasons including lip swelling (9 patients, 4.5%), mouth ulceration (11 patients, 2.6%) and stomatitis (10 patients, 2.3%). 2
It is important to understand the factors affecting safety/tolerability and the likelihood of retention on medication when making treatment decisions in clinical practice. This report therefore presents the results of a series of post hoc analyses of Study CTH-301, which were conducted primarily to identify baseline variables affecting retention on SL-APO treatment; to further explore factors potentially impacting safety and tolerability of SL-APO, such as advanced age and the concurrent use of dopamine agonists (DAs) at baseline; and to further explore the occurrence of oropharyngeal TEAEs in patients included in the study.
Methods
Study design
This study consisted of four post hoc analyses of Study CTH-301 (ClinicalTrials.gov, NCT02542696; EudraCT, 2016-000637-43). 2 Study CTH-301 was a phase III, multicentre, non-randomised, open-label study that assessed the safety, tolerability and efficacy of SL-APO when used over the long term (⩾3 years) as an on-demand treatment for OFF episodes in patients with PD and motor fluctuations, full details of which have been published previously. 2 The study consisted of an open-label dose-optimisation phase and an open-label long-term safety phase and included both patients who had not previously participated in a study with SL-APO (defined as ‘de novo patients’; i.e. patients not previously exposed to SL-APO treatment, not de novo patients with PD) as well as ‘rollover patients’ who had completed prior SL-APO studies (CTH-201 (phase II), 5 CTH-203 (phase II), 6 CTH-300 (phase III), 1 or CTH-302 (phase III) 7 ; Table S1). 2
All patients received SL-APO, with dose determination and adjustments based on efficacy, safety and tolerability. 2 For de novo patients, SL-APO dosing was determined during a 3-week dose-optimisation phase, in which SL-APO was titrated in 5-mg increments over the dose range 10–35 mg. Rollover patients initially underwent SL-APO dose optimisation (as described for de novo patients), but following a protocol amendment, they were assigned the same SL-APO dose they received in the previous study. During the long-term safety phase, patients self-administered SL-APO at home for the treatment of up to five OFF episodes per day, with a minimum of 2 h between doses. 2 Following screening and titration, patients attended clinic visits at Weeks 4, 12, 24, 36 and 48 during the first year, and were additionally contacted by telephone at Weeks 2, 8, 18, 30 and 42 to collect safety information and changes to concomitant medication. 2 During subsequent years, patients attended clinic visits at Months 4, 8 and 12 to undergo safety assessments, and were contacted by telephone at Months 2, 6 and 10 to collect safety information and changes to concomitant medication. 2 SL-APO dose adjustments were allowed at the investigator’s discretion, 2 and patients were able to continue in the study until it was terminated by the sponsor, or until SL-APO became commercially available in their country. 2
The study was conducted in accordance with the International Council for Harmonisation (ICH) Good Clinical Practice Guidance, the Declaration of Helsinki and all applicable local law(s) and regulation(s). 2 The protocol was approved by the Institutional Review Board/Independent Ethics Committee before patient enrolment and all patients provided written informed consent prior to study participation. 2
Study population
Inclusion criteria for de novo patients were age ⩾18 years; a clinical diagnosis of PD; a clinically meaningful response to levodopa (as determined by the investigator); stage 1–3 on the modified Hoehn and Yahr scale when in ON-state; a Mini-Mental State Examination score of >25; treatment with stable doses of levodopa/carbidopa and adjunctive PD medications for ⩾4 weeks (⩾8 weeks for monoamine oxidase B inhibitors) before the first screening visit and experiencing at least one OFF episode per day, with a total daily OFF time of ⩾2 h. 2 Rollover patients were included if they completed a prior SL-APO study with no major changes in concomitant PD medications from the prior study. 2 Exclusion criteria for de novo patients included atypical or secondary parkinsonism; presence of a major psychiatric disorder; mouth cankers/sores ⩽30 days before the first screening visit; a history of clinically significant hallucinations in the past 6 months; a history of clinically significant impulse control disorder(s); previous treatment with a neurosurgical procedure for PD; intraduodenal levodopa, continuous subcutaneous apomorphine (SC-APO) infusion or SC-APO ⩽7 days before the second screening visit and current treatment with 5-hydroxy tryptophan (serotonin) receptor antagonists, dopamine receptor antagonists (excluding quetiapine and clozapine) or dopamine-depleting agents. 2 Rollover patients were excluded if they developed mouth cankers/sores ⩽14 days after completing their previous SL-APO study. 2
Study assessments
The present study comprised four separate post hoc analyses. The first three analyses were conducted only in de novo patients (to eliminate bias from prior experience with SL-APO), while the fourth analysis (related to oropharyngeal TEAEs) was conducted in both de novo and rollover patients.
In the first analysis, data from Study CTH-301 were used to build a model to identify baseline variables that differ between patients who completed the study (‘completers’) and those who discontinued during the dose-optimisation or long-term safety phase (‘non-completers’) due to either lack of efficacy or adverse events (AEs). In the second analysis, safety/tolerability was compared between the subgroups of patients who were and were not receiving concurrent DA treatment at baseline (‘DA users’ and ‘non-DA users’, respectively). In addition, safety/tolerability was compared for the subgroups of non-DA users who had previously received treatment with DAs (‘pre-DA users’) and those who had never previously been treated with DAs (‘non-pre-DA users’). Safety/tolerability was assessed by evaluating the incidence of TEAEs, discontinuation due to TEAEs, DA-related TEAEs and time to discontinuation due to TEAEs. DA-related TEAEs were defined by the authors, including AEs such as nausea, dizziness, somnolence, vomiting orthostatic hypotension and dyskinesia (Table S2). Mean SL-APO doses were also compared between subgroups. In the third analysis, safety/tolerability was evaluated in the subgroup of patients aged <70 and in the subgroup aged ⩾70 years at baseline. The rationale for using this age cut-off was that the ⩾70 years cut-off provided a larger sample size than, for example, ⩾75 years. Safety/tolerability was assessed by evaluating the incidence of TEAEs, discontinuation due to TEAEs and time to discontinuation due to TEAEs. Mean SL-APO doses were also compared between subgroups. The fourth analysis examined the occurrence of oropharyngeal TEAEs in all patients (de novo and rollover) included in Study CTH-301. The incidence, severity and time to onset of oropharyngeal TEAEs and discontinuations due to oropharyngeal TEAEs were evaluated. The progression of oropharyngeal TEAEs across four initial states (no TEAE, mild, moderate, severe) to three subsequent states (mild, moderate, severe) was assessed during the long-term safety phase, considering the most severe status observed for any oropharyngeal TEAE in each patient. In cases of resolved oropharyngeal AEs, the action taken with the study drug was classified into one of the following categories: drug interruption, drug withdrawal, dose reduction or no action. Drug interruption was defined as a temporary suspension of the study drug. The differences in the proportion of patients reporting oropharyngeal TEAEs between subpopulations defined by SL-APO total daily dose (higher vs lower) and frequency of daily SL-APO intakes (higher vs lower) were examined during the long-term safety phase. The severity of oropharyngeal TEAEs was also analysed in relation to the total daily dose and frequency of daily intakes. Due to the limited number of severe TEAEs, moderate and severe cases were grouped into a single category (‘not mild’). Each patient was classified based on the maximum severity of oropharyngeal TEAEs reported during the study.
Statistical methodology
In the first analysis, the aim was to identify the baseline characteristics affecting SL-APO retention (the full list of initial baseline variables used can be found in Table S3) by building one model focusing on completers and non-completers due to lack of efficacy, and another on completers and non-completers due to AEs. A logistic regression classification algorithm with LASSO regularisation was used to identify these variables using the following steps: (1) data preparation: zero and near-zero variance predictors were removed, one-hot encoding was applied to turn categorical data into a binary numerical format, and standardisation was then applied; (2) tuning the regularisation parameter (λ) of the model: a 10-fold cross-validation was employed to identify the optimal λ, and observations were weighted by frequency to balance class distribution, addressing imbalanced outcome to identify the optimal λ; (3) model fitting and variable selection: the final logistic LASSO model was fitted into the data, and variables with non-zero coefficients were selected; (4) model performance evaluation: model performance was assessed using a 5-fold cross-validation, with stratification by class to preserve the class distribution in each fold; (5) selected baseline variables were tested for statistically significant differences between completers and non-completers using an appropriate test (two-sample t test (normally distributed variables) or Wilcoxon rank-sum test (non-normally distributed variables) for continuous variables, Chi-squared test of proportions for binary variables; two-sided p-value were calculated to determine statistical significance, with α = 0.05).
In the second and third analyses, descriptive statistics were used to summarise the incidence of TEAEs. In the second analysis, rates of discontinuation due to TEAEs and DA-related TEAEs were compared between non-DA users and DA users using the Chi-square test, and time to discontinuation due to TEAEs was compared between the two subgroups using the Wilcoxon rank sum test. When comparing SL-APO doses between the subgroups, the p-value calculated was not based on mean values but on the frequency of the doses in the two subgroups, applying the Chi-square test of independence. In the third analysis, rates of discontinuation due to TEAEs were compared between patients aged <70 years and those aged ⩾70 years old using the Chi-square test, and time to discontinuation due to TEAEs was compared between the two subgroups using the Wilcoxon rank sum test. P-values were not adjusted for multiplicity. In the fourth analysis, the incidence of oropharyngeal TEAEs was summarised using descriptive statistics. The differences in the proportion of patients reporting oropharyngeal TEAEs between subgroups defined by SL-APO total daily dose (higher vs lower) and frequency of daily SL-APO intakes (higher vs lower) were assessed by dividing patients into two groups based on the median value of each variable. A Chi-squared test for equality of proportions (two-sided) was used to evaluate these relationships, with a confidence level of 95% (α = 0.05). In addition, the Wilcoxon signed-rank test was used to evaluate the differences in both the total daily dose and the frequency of daily intakes in relation to the maximum severity of oropharyngeal TEAEs (‘mild’ vs ‘not mild’) reported by the patients. This test compared the median values for total daily dose or the frequency of daily intakes between the ‘mild’ and ‘not mild’ groups.
Results
Study population
Of the 496 patients who entered Study CTH-301 (de novo, n = 369; rollover, n = 127), 449 (90.5%) entered the dose-titration phase (de novo, n = 369; rollover, n = 80) and 47 (9.5%) were rollover patients that were assigned the same SL-APO dose they received in the previous study. 2 A total of 426/496 (85.9%) patients continued into the long-term safety phase (de novo, n = 305; rollover, n = 121), and 120/496 (24.2%) completed the long-term safety phase (de novo, n = 80; rollover, n = 40). 2 The most common reasons for discontinuation from the study were AEs (33.7% (de novo, 36.0%; rollover, 26.8%)), withdrawal of consent (21.0% (de novo, 21.4%; rollover, 19.7%)), termination of the study by the sponsor (7.3% (de novo, 5.4%; rollover, 12.6%)) and lack of efficacy (5.2% (de novo, 6.2%; rollover, 2.4%)). 2 Patients who entered the long-term safety phase remained on their optimised SL-APO dose for a median of 169.0 days (25th, 75th percentile: 82.0, 437.8).
The mean age of the patients entering the study (de novo and rollover) was 64.4 years, the mean time since PD diagnosis was 8.7 years and the mean time since the onset of motor fluctuations was 4.5 years (Table 1). The most common types of OFF episodes were wearing-off (96.8%), delayed ON (69.2%) and morning akinesia (64.9%). The mean number of OFF episodes per day at baseline was 3.9 with a mean duration of 75.3 min. Concomitant DAs were used by 315 (63.5%) patients at baseline. The most commonly used DAs (⩾10% of patients) were pramipexole (n = 120; 24.2%), ropinirole (n = 109; 22.0%) and rotigotine (n = 86; 17.3%). The demographic and baseline characteristics of the de novo patients were similar to those of the overall population (Table 1).
Patient demographic and baseline characteristics (including use of concomitant PD medications) in patients entering the study.

N = 496 for total population unless otherwise stated.
N = 494.
N = 481.
Concomitant PD medications were those with a start or stop date on or after the first date of study drug dosing.
⩾10% of de novo or roll-over patients.
max, maximum; MDS-UPDRS, Movement Disorder Society-sponsored Unified Parkinson’s Disease Rating Scale; min, minimum; N/A, not applicable; PD, Parkinson’s disease; SD, standard deviation.
Baseline factors that differed significantly between completers and non-completers
Baseline variables associated with discontinuation due to lack of efficacy
The number of daily levodopa intakes, total daily levodopa dose at baseline and presence of morning akinesia were found to differ significantly between patients who were completers and those who were non-completers due to lack of efficacy (Figure 1). Compared with non-completers due to lack of efficacy, completers had a lower number of daily levodopa intakes (p = 0.0047), a lower total daily dose of levodopa (p = 0.0012) and a higher rate of morning akinesia (p = 0.0270).
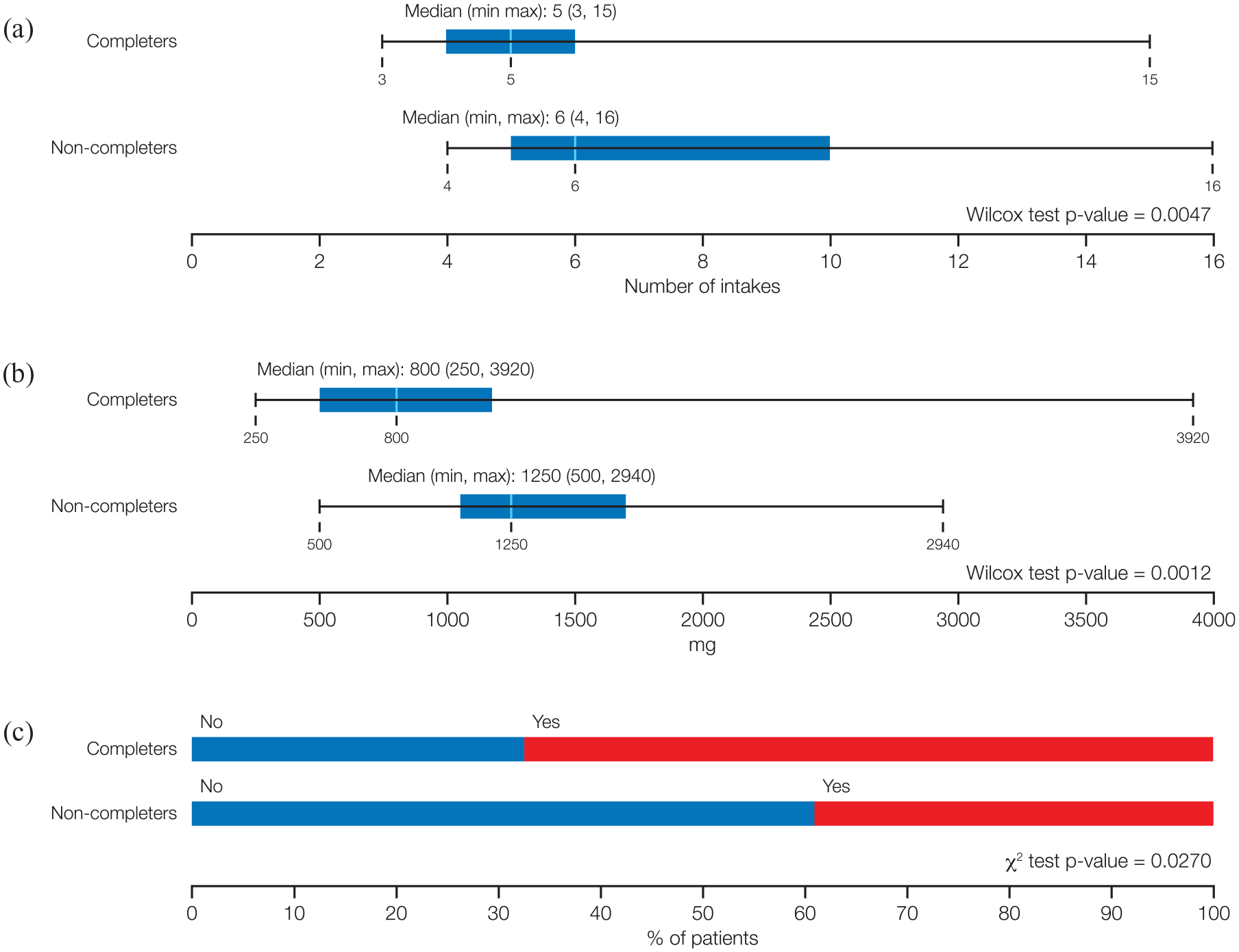
Baseline variables that differed significantly between the subgroups of patients who completed the study and those who discontinued due to lack of efficacy. (a) Number of daily levodopa intakes. (b) Total daily levodopa dose at baseline. (c) Presence of morning akinesia.
Baseline variables associated with discontinuation due to AEs
The use of DAs, age and the presence of delayed ON were found to differ significantly between patients who were completers and those who were non-completers due to AEs (Figure 2). Compared with non-completers due to AEs, completers had a higher rate of concomitant DA use (p = 0.0002), and a higher rate of delayed ON (p = 0.0448). Completers were also on average younger than non-completers (p = 0.0276; Figure 2).

Baseline variables that differed significantly between the subgroups of patients who completed the study and those who discontinued due to adverse events. (a) Dopamine agonists use. (b) Age. (c) Presence of delayed ON.
Comparison of safety and tolerability outcomes between DA users and non-DA users
Of the 369 de novo patients included in Study CTH-301, 223 (60.4%) were DA users and 146 (39.6%) were non-DA users at baseline. Compared with non-DA users, DA users demonstrated a lower incidence of the most common TEAEs (>5%) during both phases of the study (Table 2). The rates of discontinuation due to TEAEs were significantly lower in DA users versus non-DA users during the dose-optimisation phase (p < 0.001), with a trend towards statistical significance during the long-term safety phase (p = 0.089; Table 2). The rate of DA-related TEAEs was significantly lower in DA users versus non-DA users during both phases of the study (p < 0.0001 for both; Table 2). The median time to discontinuation due to TEAEs was longer in DA users versus non-DA users, although the difference was not statistically significant (148.0 vs 114.0 days; p = 0.093). The mean SL-APO optimised dose was significantly lower for non-DA users than for DA users (18.0 vs 21.2 mg; p < 0.01).
Summary of safety/tolerability in non-DA users and DA users during the dose-optimisation and long-term safety phases.
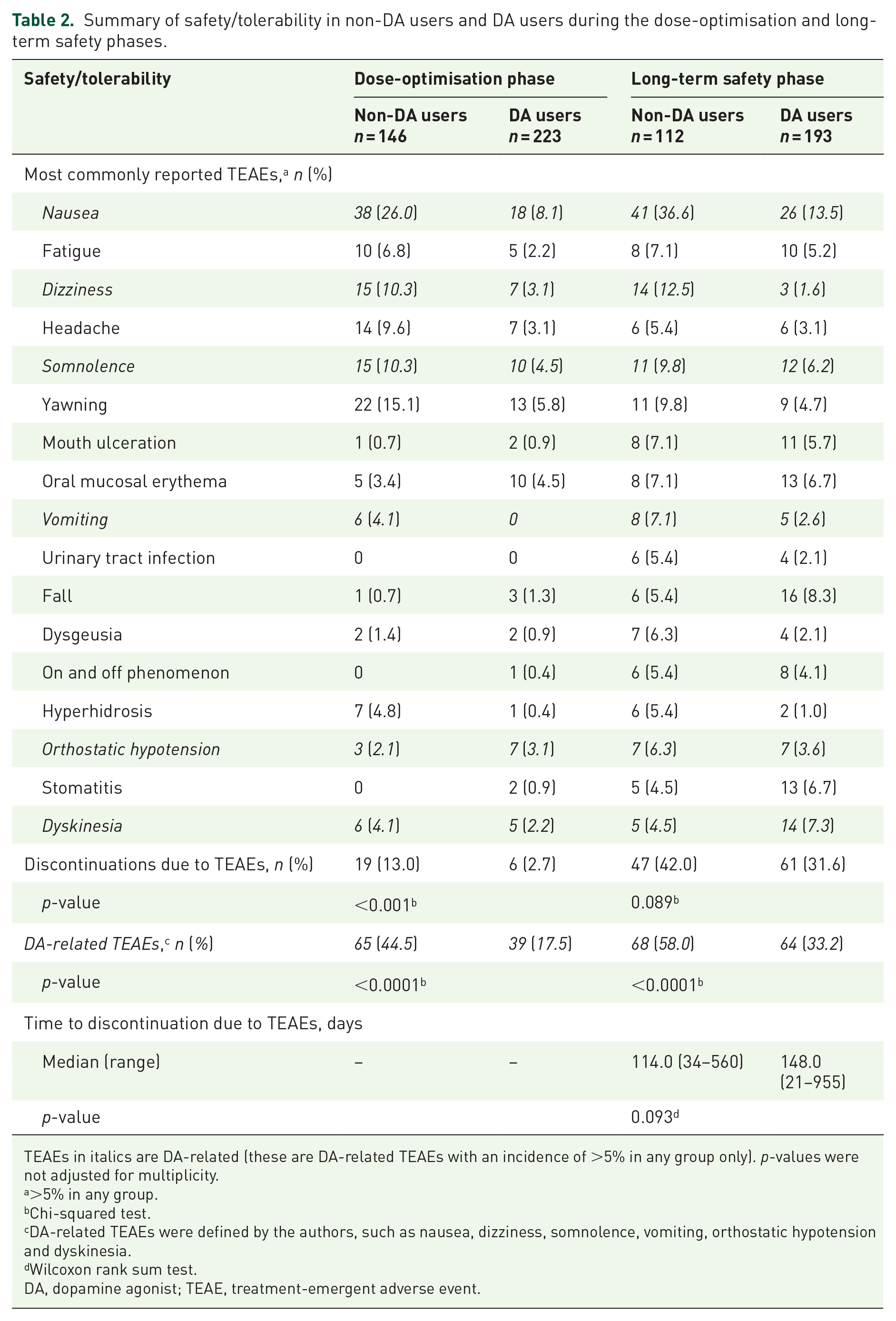
TEAEs in italics are DA-related (these are DA-related TEAEs with an incidence of >5% in any group only). p-values were not adjusted for multiplicity.
>5% in any group.
Chi-squared test.
DA-related TEAEs were defined by the authors, such as nausea, dizziness, somnolence, vomiting, orthostatic hypotension and dyskinesia.
Wilcoxon rank sum test.
DA, dopamine agonist; TEAE, treatment-emergent adverse event.
Most individuals in the non-DA user group had not previously been treated with DAs (133 patients, 91.1%), compared to 13 patients who had prior DA exposure (pre-DA, 8.9%). Discontinuation rates were similar between both groups (87.2% for non-pre-DA users; 92.3% for pre-DA users), with a similar percentage of patients discontinuing due to AEs (51.7% for non-pre-DA users; 50.0% for pre-DA users).
Comparison of safety/tolerability outcomes according to age
Of the 369 de novo patients included in Study CTH-301, 253 were aged <70 years and 116 were aged ⩾70 years. During the two study phases combined, the overall rate of TEAEs was 89.7% in both groups, with a slightly higher incidence of treatment-related TEAEs (76.7% vs 70.0%) and serious TEAEs (12.1% vs 8.7%) in the subgroup aged ⩾70 years than in the subgroup aged <70 years (Table 3). The incidence of the most common (>5%) TEAEs was generally comparable between the two age groups (Table 3), with lip swelling, orthostatic hypotension, urinary tract infection and oral paraesthesia being more commonly reported in the older than in the younger age group during the long-term safety phase. The percentage of patients discontinuing due to AEs was higher in the subgroup aged ⩾70 years than in the subgroup aged <70 years (41.4% vs 33.6%), but the difference was not statistically significant (p = 0.1839).
Summary of safety in the <70 years and ⩾70 years age groups.

AE, adverse event; TEAE, treatment-emergent adverse event.
Oropharyngeal TEAEs
Occurrence of oropharyngeal TEAEs
During both phases of the study, 206 (41.5%) patients experienced oropharyngeal TEAEs, and 81 (16.3%) discontinued due to oropharyngeal TEAEs (Table 4). During the dose-optimisation phase, 53 (11.8%) patients experienced oropharyngeal TEAEs and one (0.2%) patient discontinued due to oropharyngeal TEAEs. During the long-term safety phase, 178 (41.8%) patients experienced oropharyngeal TEAEs and 80 (18.8%) patients discontinued due to oropharyngeal TEAEs. The majority of oropharyngeal TEAEs overall and those leading to discontinuation were mild or moderate in intensity (Table 4), with more severe TEAEs being associated with higher discontinuation rates.
Summary of patients reporting oropharyngeal TEAEs during the dose-optimisation and long-term safety phases.

Based on the highest severity level for each patient.
TEAEs leading to drug discontinuation in ⩾2% of patients.
TEAE, treatment-emergent adverse event.
The most frequently reported oropharyngeal TEAEs leading to drug discontinuation during the long-term safety phase were lip swelling, mouth ulceration and stomatitis (Table 4). The median time to onset for the first oropharyngeal TEAEs experienced by the patients was 89.5 days (Figure 3). The median time to discontinuation in patients who discontinued due to oropharyngeal TEAEs was 147 days (interquartile range, 87–201 days).

Distribution of days to onset of the first oropharyngeal TEAEs during the dose-optimisation phase and long-term safety phase. Black lines indicate smoothed density estimate of the histograms; turquoise vertical lines on x-axes indicate days to onset of oropharyngeal TEAEs for each individual patient; dashed red line indicates the median; continuous red line indicates the mean.
Progression of oropharyngeal TEAEs during the long-term safety phase
During the long-term safety phase, a total of 394 drug-related oropharyngeal TEAEs were reported (Table 4); of these, 91.6% (n = 361) resolved. Of the resolved events, 3.0% (n = 12) were associated with SL-APO dose reduction, 18.3% (n = 72) with drug interruption and 33.8% (n = 133) resolved without any action taken on SL-APO; additionally, 30.7% (n = 121) required permanent drug withdrawal. The median time to resolution of drug-related oropharyngeal TEAEs not leading to drug withdrawal was 12 days (interquartile range, 3–31.8 days). Among those patients who started without these TEAEs but experienced drug-related oropharyngeal TEAEs during the long-term safety phase, most cases progressed to either mild (63.8%) or moderate (34.4%) based on the overall oropharyngeal TEAE profile of an individual patient (Table 5). In the majority of patients with mild or moderate events, no worsening in the severity of the events was reported (66.7% in both cases). Patients who initially experienced severe drug-related oropharyngeal TEAEs transitioned to mild or moderate TEAEs. Progression from no TEAE, mild TEAE or moderate TEAE into the severe state was relatively low (1.8%, 1.2% and 3.3%, respectively).
Progression of patients from no/milder to more advanced oropharyngeal TEAE severity during the long-term safety phase.

The states refer to the overall oropharyngeal TEAE profile of an individual patient. The severity of the state is determined by the most severe status observed during the study period.
TEAE, treatment-related adverse event.
Relationship between oropharyngeal TEAEs and SL-APO daily dose or intake frequency
The mean total daily dose of SL-APO was higher in patients reporting oropharyngeal TEAEs than in those not reporting oropharyngeal TEAEs (38.3 vs 32.1 mg/day; p = 0.024). During the long-term safety phase, patients using total daily doses of SL-APO above the median value (28.1 mg) showed a higher frequency of oropharyngeal TEAEs than patients with total daily doses below the median (41.2% vs 27.7%; p = 0.01; Figure 4). Similarly, the frequency of oropharyngeal events was higher in the group of patients using a number of daily intakes above the median (1.5 intakes per day) than in patients using fewer intakes (40.8% vs 28.2%; p = 0.02; Figure 4). The mean total daily dose of SL-APO was strongly correlated with the mean number of daily intakes of SL-APO (r2 = 0.723).

Frequency of oropharyngeal AEs by SL-APO total daily dose and number of intakes during the long-term safety phase. Patients were grouped based on their use of SL-APO. (a) Mean total daily dose above or below the median value (28.1 mg) or (b) mean number of daily intakes above or below the median value (1.5 intakes). Red colour indicates the percentage of patients reporting oropharyngeal AEs; blue indicates the percentage of patients without oropharyngeal AEs.
Patients with moderate or severe oropharyngeal TEAEs used a higher mean total daily dose (per diary) of SL-APO than patients with mild oropharyngeal TEAEs (42.5 (23.7) vs 33.9 (26.4) mg; p = 0.024). Similarly, patients with moderate or severe oropharyngeal TEAEs used a higher mean number of SL-APO intakes per day than those with mild oropharyngeal TEAEs (2.2 (1.2) vs 1.7 (1.2) mg; p = 0.010).
Discussion
These post hoc analyses of data from Study CTH-301 provide insights into factors affecting retention on SL-APO and its safety/tolerability profile in different subgroups. In Study CTH-301, discontinuation rates for de novo patients were 17% and 61% in the dose-titration and long-term safety phase, respectively, with AEs being the most common reason for discontinuation during both phases (7% and 29%, respectively). 2 Several baseline factors were identified that significantly affected the likelihood of continued SL-APO use. Patients who discontinued SL-APO due to lack of efficacy were using significantly more levodopa at baseline (in terms of both the total daily dose of levodopa and the number of daily intakes) and had a significantly lower rate of morning akinesia at baseline than those who completed the study or discontinued for other reasons. Patients who discontinued SL-APO due to AEs were on average older and had a significantly lower rate of using concomitant DAs and, statistically, were associated with a lower rate of delayed ON at baseline.
The management of OFF episodes in individuals with PD requires a tailored approach for each patient.8,9 The findings of the current study have the potential to help clinicians identify the types of patients who are most likely to benefit from SL-APO as an on-demand treatment for OFF episodes in terms of patient characteristics, levodopa usage, current treatment and OFF symptoms. Indeed, those in a younger age group using less levodopa and taking concomitant DAs are likely to particularly benefit from SL-APO therapy. It is plausible that individuals requiring less levodopa are at earlier stages of the disease or have less severe symptoms,10,11 allowing SL-APO to meet a higher proportion of their therapeutic needs compared to those in more advanced stages. From a disease perspective, our results indicate that patients experiencing morning akinesia, and those who experience delayed ON might also particularly benefit from SL-APO, as previously proposed by PD experts in several publications.12–14
The significantly lower discontinuation rate due to AEs among individuals using concomitant DAs at baseline could suggest that prior exposure to DAs may help acclimatise patients to dopaminergic side effects. This is further supported by the results of the second analysis, which demonstrated that SL-APO was better tolerated in patients using DAs at baseline (DA users) than in non-DA users and the rate of DA-associated TEAEs was higher in non-DA users than in DA users. Our observations are consistent with findings from studies showing that prior exposure to dopaminergic drugs can lead to increased tolerability. One study demonstrated that patients with PD previously treated with levodopa exhibited less apomorphine-induced orthostatic hypotension compared to untreated patients, indicating increased tolerability. 15 Similarly, another study found that repeated apomorphine injections led to reduced incidence of nausea and hyperthermia in normal volunteers, 16 further supporting the notion that repeated dopaminergic exposure may mitigate peripheral side effects.
To further clarify the impact of prior DA use on SL-APO safety/tolerability profile, we assessed discontinuation rates (for any reason and due to AEs) in individuals in the non-DA users’ group who had previously been treated with DAs (‘pre-DA users’), compared with those who had never been treated with DAs before starting SL-APO (‘non-pre-DA users’). The rates of discontinuation were similar in both groups, suggesting that prior, interrupted exposure to DAs may not necessarily lead to greater tolerability of SL-APO. However, the number of patients with prior DA exposure was very low and the duration of time between stopping prior DA therapy and starting SL-APO treatment was not known in this subgroup, limiting our ability to draw conclusions. The finding that the mean daily SL-APO dose was significantly lower for non-DA users than for DA users is somewhat puzzling but might indicate that non-DA users were more cautious about using SL-APO than DA users. This could be due to patients becoming less sensitive to SL-APO, the more they are exposed to the DAs. 17 Cells chronically exposed to a strong agonist may require higher doses of the agonist to achieve the same level of excitation/activation over time.18–20
Overall, older patients were more likely to discontinue SL-APO due to AEs. These findings are not unexpected as elderly patients tend to report more AEs 21 due to age-related changes in pharmacokinetics and pharmacodynamics, increased burden of comorbidity and greater use of polypharmacy. 22 More specifically, hyposalivation and xerostomia are common in patients with PD,23,24 and age is an independent risk factor for the development of xerostomia. 25 These factors are all likely to affect the tolerability of a sublingual medication. Nevertheless, similar rates of TEAEs between the groups of patients aged <70 or ⩾70 years suggest that SL-APO has a good tolerability profile even in elderly patients. Although clinicians should be alert for the development of adverse reactions in all patients, the current findings indicate that increased vigilance may be warranted in those not currently using other DAs and the most elderly.
In the overall study, around 40% of the patients experienced oropharyngeal TEAEs and approximately half of them discontinued due to these events. The current analysis indicates that oropharyngeal TEAEs, including those leading to discontinuation, were mostly mild or moderate, predominantly occurred within the first few months of SL-APO initiation and resolved within a median of 12 days for those who did not discontinue the medication. Severe oropharyngeal TEAEs were rare and generally transient, often transitioning into less severe states. In general, oropharyngeal TEAEs resolved without the need for drug withdrawal or dose reduction.
The increased usage of SL-APO (i.e. total daily dose or number of intakes) was associated with a higher risk of developing oropharyngeal TEAEs. Similarly, individuals who experienced moderate or severe oropharyngeal TEAEs received on average a higher daily dose and/or higher number of daily intakes than those who experienced mild events. Importantly, no relationship has been observed between oropharyngeal AEs and the optimised dose (i.e. 10–30 mg) of SL-APO. 1 Oropharyngeal TEAEs leading to drug discontinuation during the long-term safety phase included lip swelling, mouth ulceration and stomatitis. An additional analysis identified two baseline characteristics significantly associated with discontinuation due to oropharyngeal TEAEs: older age and the use of DAs (data not shown). 26 While the association between discontinuations and older age aligns with the findings in this report, the higher discontinuation rate in DA-users was unexpected, but it may reflect the higher optimised doses and/or more frequent intakes of SL-APO in this subgroup of patients, leading to increased exposure. 26
Although it is unclear why mild events led to discontinuation, this perhaps reflects subjective differences in experiencing oropharyngeal side effects and how easy or difficult these are to tolerate. It should also be considered that patients might experience more than one oropharyngeal TEAE which might have decreased their tolerability of SL-APO. To minimise the risk of oropharyngeal TEAEs, it may be helpful to maintain good mouth hygiene and advise patients to rinse with water before and after intakes. This practice could help restore the mucosa’s pH, reduce irritation and remove any residual product. 27 According to the product summary characteristics, SL-APO is not recommended to be reintroduced following discontinuation due to oropharyngeal TEAEs, since such adverse reactions may recur with increased severity. 3
ON and OFF periods tend to become unpredictable over time and ‘on demand’ therapies for OFF episodes that can be adapted to patients’ needs and symptoms are increasingly being used to manage these episodes. 28 For these reasons, various formulations of antiparkinsonian medications are being explored and a few studies have compared SL-APO with other apomorphine formulations in terms of efficacy and safety.6,7,29,30
In a randomised, crossover, open-label study comparing the pharmacokinetics and comparative bioavailability of a single dose of SL-APO with two SC-APO formulations (SC-APO (APOKYN® approved in the USA) and SC-APO-GO (APO-go PEN® approved in Europe), both administered by a multiple-dose pen injector), safety findings were similar across the three formulations. 6 In a multicentre, open-label, randomised, crossover study of SL-APO versus SC-APO, both formulations were well tolerated with similar safety profiles. 7 The frequencies of AEs leading to discontinuation were comparable between the two formulations during the dose-optimisation phase (SL-APO, 3.9%; SC-APO, 5.2%) and the treatment phase (SL-APO, 5.6%; SC-APO, 2.9%) and most AEs were mild or moderate. 7 However, patients reported an overall preference for and greater satisfaction with SL-APO, compared with SC-APO, 7 as also indicated by a discrete choice experiment, in which 300 US participants with PD expressed a preference for a theoretical dissolvable sublingual film over theoretical inhaled or injected medicines, regardless of the presence of AEs. 29
Finally, an indirect comparison of SL-APO and levodopa inhalation powder (CVT-301) was conducted, in which patient-level data from the phase III SL-APO CTH-300 trial 1 were re-weighted to match average baseline characteristics from patients included in a phase III, CVT-301 trial (SPAN-PD).30,31 The comparison study did not report data on safety but noted that SL-APO use may be limited by oral/pharyngeal soft tissue swelling, pain and paraesthesia, while the use of CVT-301 was restricted by cough and upper respiratory tract infections. Taken together, these studies indicate that SL-APO has an acceptable safety profile and is as well tolerated as other on-demand apomorphine formulations, but its sublingual method of delivery may offer increased convenience for patients. Convenience is further enhanced by the ability to self-optimise SL-APO dosing at home. 4
Study CTH-301 has several limitations that have been discussed elsewhere 2 but are also relevant to the current analyses; being the high rate of discontinuation due to withdrawal of consent one of them (21.0% overall; 21.4% in de novo patients and 19.7% in rollover patients). Although the reasons for this are unclear, it is possible that some of those who withdrew consent did so due to a perceived lack of efficacy; it is also worth bearing in mind that the study duration was ⩾3 years. The current analyses were also limited in being post hoc and not predefined. Given that these post hoc analyses were not pre-specified in the original protocol, the findings are exploratory in nature and should be interpreted accordingly. The sample size for some patient subgroups (e.g. the pre-DA users’ subgroup) was small, and the study was not powered for the statistical tests employed.
Conclusion
In summary, the findings of these post hoc analyses of Study CTH-301 identified patient characteristics associated with increased retention on SL-APO and provided insights into the safety and tolerability profile of SL-APO, which together may help clinicians and patients make informed decisions when selecting a potential on-demand treatment for OFF episodes in PD. Patients in a younger age group, those experiencing morning akinesia or delayed ON and those receiving lower dose and fewer intakes of levodopa and taking concomitant DAs are more likely to benefit from SL-APO treatment. In addition, patients not receiving concomitant DAs, those in an older age group and those taking higher dose or more frequent SL-APO intakes should be more closely monitored for general AEs, DA-related AEs and oropharyngeal AEs. The study also provides further information on the most common type of side effects associated with SL-APO treatment – namely, oropharyngeal TEAEs – which may allow their occurrence to be mitigated (e.g. in terms of daily dose and number of daily intakes) and help inform patients about what to expect when starting SL-APO treatment.
Supplemental Material
sj-doc-1-tan-10.1177_17562864251343583 – Supplemental material for Insights into retention and safety/tolerability of apomorphine sublingual film in patients with Parkinson’s disease and OFF episodes: post hoc analyses of a phase III, open-label study
Supplemental material, sj-doc-1-tan-10.1177_17562864251343583 for Insights into retention and safety/tolerability of apomorphine sublingual film in patients with Parkinson’s disease and OFF episodes: post hoc analyses of a phase III, open-label study by Jan Kassubek, Diego Santos Garcia, Wolfgang H. Jost, Lars Wojtecki, Fradique Moreira, Miguel M. Fonseca, Glynn Harrison-Jones, Isabel Pijuan and Carmen Denecke Muhr in Therapeutic Advances in Neurological Disorders
Supplemental Material
sj-docx-2-tan-10.1177_17562864251343583 – Supplemental material for Insights into retention and safety/tolerability of apomorphine sublingual film in patients with Parkinson’s disease and OFF episodes: post hoc analyses of a phase III, open-label study
Supplemental material, sj-docx-2-tan-10.1177_17562864251343583 for Insights into retention and safety/tolerability of apomorphine sublingual film in patients with Parkinson’s disease and OFF episodes: post hoc analyses of a phase III, open-label study by Jan Kassubek, Diego Santos Garcia, Wolfgang H. Jost, Lars Wojtecki, Fradique Moreira, Miguel M. Fonseca, Glynn Harrison-Jones, Isabel Pijuan and Carmen Denecke Muhr in Therapeutic Advances in Neurological Disorders
Footnotes
Appendix
Acknowledgements
We would like to thank Sumitomo Pharma for conceding the data and approving the publication. Bial – Portela & Cª conducted the analyses presented in this study with the authorisation and approval of Sumitomo Pharma. Editorial assistance was provided by John Scopes and Eliana D’Araio of mXm Medical Communications.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.