
Abstract
Background:
Changes in the original protocol of clinical trials should be clearly declared to the readers of journal publications. Otherwise, they can lead to selective outcome reporting bias or distort the appropriate judgement of the study’s external validity and statistical power, among other problems.
Objectives:
To identify silent protocol modifications in phase III and IV clinical trials examining multiple sclerosis (MS) drugs that have been carried out between 2010 and mid-2023.
Design:
Comparative analysis of ClinicalTrials.gov and associated peer-reviewed journal publications.
Methods:
An advanced search in ClinicalTrials.gov was performed and consecutive searches in PubMed, EMBASE and Google Scholar were conducted looking for the main journal publication derived from each trial. Information regarding trial design, eligibility criteria, primary outcomes and sample size estimation was simultaneously collected from ClinicalTrials.gov and publications, and subsequently compared.
Results:
In total, 112 trials were appraised. Most studies matched between data sources in terms of study arms (96.4%), assignment (99.1%) and randomization (100.0%). Concordance was also high but comparatively lower for masking (82.1%). A total of 3051 eligibility criteria were extracted, 45.5% of which matched, 25.1% were omitted in publications, 2.8% were modified and 26.6% were added. Fifty-eight trials (51.8%) completely matched regarding their published primary outcomes, whereas 20 had major inconsistencies (17.9%) and 34 (30.4%) minor inconsistencies. Fourteen trials were inconsistent in their estimated sample size; among these, the median difference between registry and publications was 36.5 individuals (interquartile range 17–161). The proportion of trials exhibiting silent protocol changes was similar regardless of study phase, industry involvement or type of registration.
Conclusion:
Silent protocol changes are common in MS clinical trials and potentially hinder the interpretation and applicability of results. Efforts must be made to promote more transparency in the field of MS clinical research.
Keywords
Introduction
Clinical trials are the cornerstone of evidence-based medicine, providing the rational basis that guides physicians in their decision-making. 1 Transparency and accountability in their planning, conduction and reporting is an intrinsic public health good and an ethical imperative. 2 Peer-reviewed journals are the gold-standard for communicating clinical trials to the medical community, 3 allowing the full scrutiny and reproduction of their methods and results. However, empirical observations indicate that a substantial proportion of clinical trials remains unpublished or is published only after major delays. 4 Moreover, trials with positive results might be more likely to be published (the so-called ‘publication bias’), thus distorting the available evidence establishing the safety and efficacy of novel drugs. 5
In addition to these issues, it has also been emphasized that methods reported in journal publications do not always adhere to the study protocol, including variations in eligibility criteria, primary outcome measures and sample size estimations, among others. 4 These methodological discrepancies can involve silent protocol modifications, which are not explicitly disclosed to the journal reader but nonetheless can have significant consequences. For instance, there could be selective outcome reporting bias, 6 which means that only the subset of pre-specified outcome measures that have positive results are chosen for publication or are reported as the study primary outcomes. Similarly, undisclosed changes in eligibility criteria can hinder the appropriate judgement of the trial’s external validity. 7 For example, the omission of pre-specified exclusion criteria in the journal publication could lead to the erroneous assumption that the trial results are more widely applicable.
To counteract all these problems, regulatory 8 and editorial authorities9,10 have increasingly required the use of clinical trial registries. The largest by far is ClinicalTrials.gov, with more than 530,000 registered studies from 229 countries and territories as of March 2025. 11 ClinicalTrials.gov summarizes key protocol details and other information to support the enrolment and tracking of clinical trials, and also allows researchers to post the trial results after their completion. 12 Moreover, the website includes a ‘history of changes’ function that enables users to navigate the archive site and compare the original protocol summary with the final version, thus assessing how closely the trial followed what was originally planned. Consequently, ClinicalTrials.gov has been used by other authors to identify protocol changes in trials coming from different fields, including surgery, 13 oncology, 14 haematology, 15 paediatrics 16 and orthodontics. 17
Multiple sclerosis (MS) is a neurological condition for which many treatments have been approved in recent years, 18 including 13 disease-modifying therapies (DMTs) (peg-interferon beta-1a, teriflunomide, dimethyl fumarate, diroximel fumarate, monomethyl fumarate, fingolimod, ozanimod, ponesimod, siponimod, cladribine, ocrelizumab, ofatumumab and ublituximab) and two specific symptomatic drugs (fampridine and delta-9-tetrahydrocannabinol/cannabidiol) authorized by the Food and Drug Administration (FDA) or the European Medicines Agency (EMA) since 2010. Therefore, studies focusing on the quality and transparency of clinical trials that led to their approval are particularly relevant. In a previous publication, we demonstrated that clinical trials evaluating drugs for MS are prone to publication bias. 19 In the current study, we aimed to identify silent protocol modifications in MS clinical trials by comparing the original information reported in ClinicalTrials.gov with what was ultimately published in peer-reviewed journals.
Materials and methods
Data source and search strategy
A search was performed on November 1, 2023 in ClinicalTrials.gov to identify clinical trials carried out between 2010 and mid-2023. The advanced search engine for this website was used applying the terms ‘multiple sclerosis’ and the filters ‘phase III’, ‘phase IV’, ‘start date on or after 1/1/2010’ and ‘primary completion on or before 7/1/2023’.
Clinical trial selection
The information pertaining to each trial record was reviewed, and its eligibility was established attending to the following criteria: to be included, phase III or IV clinical trials examining MS drugs should be completed and have their results available in peer-reviewed journal publications. A trial was considered to be completed if it was described as ‘completed’ or ‘terminated’ in ClinicalTrials.gov or if its primary outcome result was available at ClinicalTrials.gov or in a matching journal publication. Studies classified as ‘withdrawn’, trials not limited to MS population and studies evaluating diagnostic procedures or non-pharmacological interventions were excluded. Trials were reviewed independently by two authors, and disagreements were resolved by consensus.
Search for matching journal publications
In November 2023, consecutive searches in PubMed, EMBASE and Google Scholar were conducted to identify the main peer-reviewed publication associated with each trial. The methodology used for this purpose has been described previously. 19 Letters to the editor, interim analysis, ancillary studies, pooled studies, systematic reviews, meta-analyses and publications in languages other than English were excluded. If several publications were identified for the same study, the one reporting the study primary outcome was chosen. If no publication was found, the principal investigators and sponsors were e-mailed sequentially requesting information up to three times. The search was updated in February 2024.
Data extraction
Data were extracted using a predesigned Microsoft Excel worksheet. The following information was simultaneously collected from ClinicalTrials.gov and the corresponding journal publication (including text, tables, figures, references, supplemental material and links available on the journal website): clinical trial design (study arms, assignment, masking and randomization), eligibility criteria, primary outcome measures, estimated sample size and sponsors and collaborators. Information regarding the statistical analysis of the primary outcome was not extracted since it is not submitted to ClinicalTrials.gov when the trial is registered, but rather when the trial results are reported to the registry, thus hindering the identification of the pre-specified analyses. Registration dates were retrieved from ClinicalTrials.gov to determine if the trial had been prospectively or retrospectively registered (i.e. before or after the enrolment of the first participant). For prospectively registered trials, we used the version of the ClinicalTrials.gov’s record that was in force on the study actual start date (i.e. the version in effect when the first participant was enrolled, according to the registry). For retrospectively registered trials, we used the earliest version of the record. For this purpose, ClinicalTrial’s ‘history of changes’ function was used. Trial duration was defined as the interval between the study start date and the study’s actual primary completion date, as per ClinicalTrials.gov. Other data extracted for journal publications comprised corresponding author affiliation, where the study was conducted, the actual sample size and the primary outcome results.
Eligibility criteria were classified in seven mutually exclusive categories: ‘disease definition’, ‘demographics’, ‘comorbidities’, ‘treatments’ (previous, current or programmed use of other therapeutical interventions), ‘reproductive health’ (contraceptive methods, pregnancy and lactation), ‘legal & ethical’ (e.g. informed consent) and ‘organizational & administrative’ (e.g. anticipated non-compliance) (see examples in Supplemental Table 1).
We used the data available on Drugs@FDA 20 and the EMA’s summaries of product characteristics (SmPCs) to identify the specific clinical trials included in our study that led to the drug approval by these two regulatory agencies.
Data analysis
A descriptive analysis was conducted based on the information derived from journal publications. Qualitative variables were expressed as absolute frequency (n) and relative frequency (%). Quantitative variables were expressed as median and interquartile range (IQR). Subsequently, data from ClinicalTrials.gov was compared with that of the matching journal publications to identify protocol inconsistencies. Protocol modifications explicitly stated in the publication (or declared as amendments within the protocol when it was available as supplemental material, referenced in the publication or accessible through links on the journal’s website) were not considered inconsistencies. Inconsistencies were appraised independently by two authors, and disagreements were resolved by consensus.
Regarding clinical trial design, any lack of match was considered to be an inconsistency. Specifically for masking, it was appraised if there was a match for every study party (i.e. participants, care providers, investigators and outcome assessors). If the journal publication did not provide detailed information, the overall number of masked parties was used for comparisons between data sources.
Inconsistencies in eligibility criteria were classified as ‘omission’ (if the information specified in ClinicalTrials.gov was not reported in the publication), ‘modification’ (if it was somehow changed) or ‘addition’ (if new information was mentioned in the publication), similarly to Blümle et al. 7 Synonym expressions and equivalent criteria were considered only once (e.g. ‘relapsing forms of MS’ as inclusion criterion and ‘non-relapsing forms of MS’ as exclusion criterion). For each inconsistency in eligibility criteria, it was appraised whether it implied that the study population described in the journal was broader, narrower or similar compared to that in the registry. Lastly, an overall assessment was done grouping all eligibility criteria coming from the same clinical trial. In this analysis, populations were considered to be ‘not comparable’ if there was a mixture of inconsistencies with opposed implications (i.e. some broadening and others narrowing the study population).
Inconsistencies in primary outcome measures were classified according to an adaptation of the method used by Chan et al. 6 : ‘omission’ (the primary outcome of the registry was not reported in the publication), ‘downgrade’ (it was reported as non-primary or its hierarchy was not specified in the publication), ‘addition’ (a new primary outcome was introduced in the publication), ‘upgrade’ (a non-primary outcome was reported as primary in the publication), ‘modification’ (the definition of the outcome, such as the time frame, was changed in the publication) and ‘different completeness’ (details of the outcome definition were missing in one of the two data sources, such as the specific measurement (e.g. ‘Expanded Disability Status Scale’), the metric (e.g. ‘change from baseline’), the method of aggregation (e.g. ‘proportion of patients with a 1.5 point increase’) or the time frame (e.g. ‘after 6 months’)). The first four scenarios were considered major inconsistencies, and the last two minor inconsistencies. Similarly to the method used by Mathieu, 21 we classified the consequences of each primary outcome inconsistency as ‘positive’ or ‘negative’ based on whether the result that was omitted, downgraded, added or upgraded significantly supported the superiority (or non-inferiority/equivalence) of the experimental drug from a statistical point of view. For instance, omitting an efficacy outcome for which there are no differences between the experimental drug and placebo (p = 0.210) is classified as ‘positive effect’, since it hides an unfavourable result. Minor inconsistencies and inconsistencies involving outcomes with unavailable or ambiguous results were classified as ‘undetermined’ (Table 1).
Effect of primary outcome measure inconsistencies.

Results that statistically support the superiority, non-inferiority or equivalence of the experimental drug.
Results that do not statistically support the superiority, non-inferiority or equivalence of the experimental drug.
Results that are unavailable or whose interpretation is ambiguous from a clinical point of view (e.g. changes in lymphocyte subpopulations), irrespective of their statistical significance.
Inconsistencies in the estimated sample size were expressed as the median difference in the estimated number of participants between the registry and the publication, along with the IQR. Regarding the names of sponsors and collaborators, changes due to rebranding of companies or institutions over time were not considered to be discrepancies.
To assess if the proportion of trials showing inconsistencies varied depending on study phase, industry involvement (either sponsorship or collaboration), registration status (prospective or retrospective) or whether the trial led to the drug approval by the FDA and/or EMA (only for phase III studies), Fisher’s exact test or χ² test were used. These tests were also used to appraise if the similarity between the published study population and that described in the registry was influenced by study phase. A two-tailed p-value of less than 0.05 was considered statistically significant. Data analysis was performed using SPSS 29.0.1.0 for Mac (SPSS Inc., Chicago, IL, USA).
Results
Search process
Through the advanced search in ClinicalTrials.gov, 279 clinical trials were identified, 205 of which were completed eligible trials (Figure 1). Of these, 116 had publications available in peer-reviewed journals. Four were excluded because their associated publications were in Russian. Thus, 112 clinical trials were included in the analysis, representing 56,524 individuals.

Flowchart depicting the process to select the clinical trials included in the analysis.
Clinical trial characteristics
Table 2 describes the characteristics of the 112 clinical trials included in the analysis. These studies evaluated a total of 35 experimental drugs. Most of them were phase III (50.9%), randomized (54.5%), controlled (69.6%) clinical trials evaluating DMTs (82.1%). Seventeen specific trials led to FDA and/or EMA approving the drug or expanding its indication, and all of them were on DMTs. Sixty-four trials (57.1%) were prospectively registered in ClinicalTrials.gov. Industry was involved, either as sponsor or collaborator, in most studies (87.5%).
Description of the clinical trials included in the analysis (n = 112).
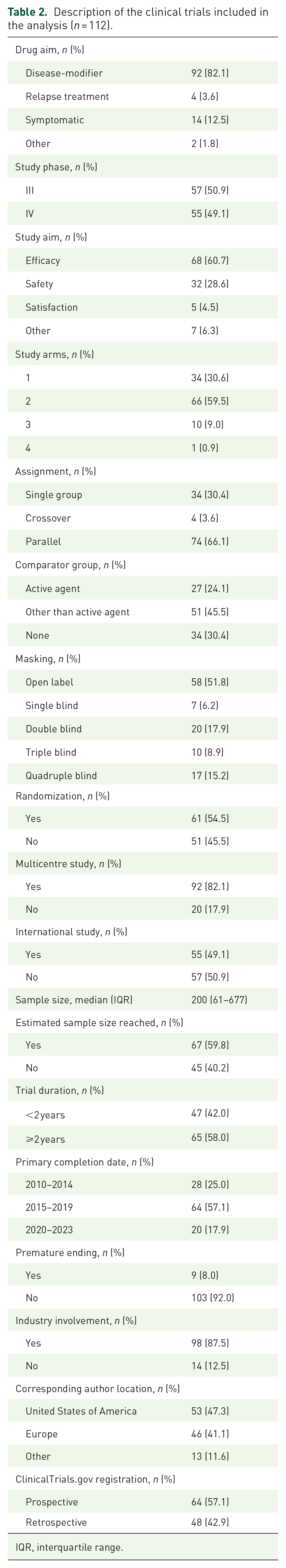
IQR, interquartile range.
Comparison between ClinicalTrials.gov and journal publications
Clinical trial design
Nearly all publications matched ClinicalTrials.gov in terms of the number of study arms (96.4%), assignment (99.1%) and randomization (100.0%). Concordance for masking was also high but comparatively lower (82.1%). Fifteen trials reported a higher level of blinding in the registry (13.4%), while five trials reporting a higher level of blinding in the publication (4.5%). In addition, 14.8% of masked trials (8/54) did not clearly describe in their publications which study parties were unaware of the intervention, only providing the overall number of parties that were blinded.
Eligibility criteria
Overall, 3051 eligibility criteria were extracted from the 112 trials. Most of them belonged to ‘comorbidities’ category (36.2%), followed by ‘treatments’ (28.4%), ‘disease definition’ (13.0%), ‘reproductive health’ (7.8%), ‘demographics’ (7.4%), ‘legal & ethical’ (3.7%) and ‘organizational & administrative’ (3.5%). The median number of reported eligibility criteria was 17 on ClinicalTrials.gov (IQR 11–26) and 16 in publications (IQR 9–32). When comparing both data sources, 45.5% of the eligibility criteria matched, 25.1% were omitted in the publication, 2.8% were modified and 26.6% were added (see Figure 2(a) for a detailed breakdown). Only two trials were fully concordant in all their eligibility criteria.

Discrepancies in eligibility criteria and their implications. (a) Comparison of registry and publication eligibility criteria (n = 3051 criteria). (b) Implications of inconsistencies in eligibility criteria (n = 1164 inconsistent criteria). (c) Similarity of registry and publication study populations (n = 112 clinical trials).
When we appraised the implications of non-matching eligibility criteria, we found that in 40.3% of the cases these inconsistencies broadened the study population in the journal publication in comparison to the registry, in 43.1% they narrowed it and in 16.6%, they did not imply a relevant change (see Figure 2(b) for a detailed breakdown; find examples in Supplemental Table 2).
When we grouped and collectively assessed all eligibility criteria pertaining to the same clinical trial, the study population was overall broader in the journal publication than in the registry in 24 trials (21.4%), it was narrower in 27 trials (24.1%), it was not comparable in 53 trials (47.3%) and it was similar in both data sources in only 8 trials (7.1%) (see Figure 2(c) for a detailed breakdown). ‘Comorbidities’ was the category for which trial populations were less often similar (21.4%), followed by ‘treatments’ (26.8%), ‘reproductive health’ (39.3%), ‘disease definition’ (50.0%), ‘demographics’ (82.1%) and ‘legal & ethical’ (100.0%) and ‘organizational & administrative’ (100.0%). There was no remarkable imbalance in any category regarding the proportion of trials broadening versus narrowing the published study population, with the exception of ‘demographics’ (17.0% vs 0.9%) and ‘reproductive health’ (37.5% vs 21.4%).
Primary outcome measures
Fifty-eight trials (51.8%) completely matched in terms of their published primary outcomes. Twenty had major inconsistencies (17.9%) and 34 (30.4%) minor but not major inconsistencies, being ‘downgrade’ and ‘different completeness’ the most common, respectively (Table 3). Thirteen primary outcome inconsistencies had a positive effect (14.4%), 10 had a negative effect (11.1%) and 67 had an undetermined effect (74.4%).
Primary outcome inconsistencies.

Note that a clinical trial can report more than one primary outcome measure.
Mutually exclusive categories.
Estimated sample size
The estimated sample size was always reported in ClinicalTrials.gov, but it was omitted in 46 journal publications (41.1%). When it was available in both data sources, it matched in 78.8% of the occasions (52/66). Nine trials reported a higher number in the registry and five trials reported a higher number in the publication. Within discrepant studies, the median difference between the estimated sample size in the registry and the estimated sample size in the publication was 36.5 individuals (IQR 17–161).
Sponsors and collaborators
Disclosure of sponsors and collaborators was concordant in 83.9% of the trials. Inconsistencies in this regard implied the omission of industry involvement in nine trials, all of which reported non-industry sponsors and collaborators in the registry.
Factors related with inconsistencies between ClinicalTrials.gov and journal publications
The proportion of trials with protocol inconsistencies in study design, eligibility criteria, primary outcome measures and estimated sample size did not vary depending on study phase (Supplemental Table 3), industry involvement (Supplemental Table 4), trial registration status (Supplemental Table 5) or whether the trial led to the drug approval (Supplemental Table 6).
The sole exception was a significantly lower proportion of phase III trials exhibiting major inconsistencies in their primary outcomes compared to phase IV trials (10.5% vs 25.5%, p = 0.039). However, the proportion of primary outcome inconsistencies leading to a positive, negative or undetermined effect did not differ between phase III and phase IV trials (positive effect 8.3% vs 18.5%, negative effect 16.7% vs 7.4%, undetermined effect 75.0% vs 74.1%, p = 0.200). Given the sample size, caution is needed when interpreting this sub-analysis.
The similarity between the published study population and that described in the registry significantly changed depending on the study phase (III vs IV) in the case of two categories: ‘demographics’ (broader in publication 8.8% vs 25.5%, narrower in publication 1.8% vs 0.0%, similar 89.5% vs 74.5%, p = 0.024) and ‘comorbidities’ (broader in publication 29.8% vs 34.5%, narrower in publication 40.4% vs 16.4%, not comparable 15.8% vs 20.0%, similar 14.0% vs 29.1%, p = 0.028).
Discussion
In this cross-sectional study, we have comprehensively examined silent protocol modifications in phase III and IV clinical trials on MS. The comparison of the original ClinicalTrials.gov’s record with the corresponding journal publications uncovered numerous unexplained methodological discrepancies related to trial design, eligibility criteria, primary outcome measures and sample size estimation that were not duly declared in the peer-reviewed publications. Our findings highlight shortcomings in accountability and transparency that should be considered by patients, clinicians, researchers and decision-makers, as these unreported protocol modifications could hinder the interpretation and applicability of MS trial results.
In our study, we found that most studies were consistent in terms of the number of study arms, assignment, randomization and masking. However, masking did not match the original protocol for nearly one-fifth of the trials (e.g. the study was stated to be open-label in ClinicalTrials.gov, but the outcome assessor was blinded according to the publication). As previously reported for medicine in general, 22 we also found that masking was poorly described in some MS publications, since they commonly used ambiguous terminology (e.g. ‘double blind’) without explicitly stating who was masked to the intervention. This lack of clarity hampers the journal reader to correctly appraise performance and detection biases, 23 which are key elements to assess study quality.
Another important finding was that half of the eligibility criteria did not match when comparing publications with ClinicalTrial.gov’s records. This discrepancy often implied that the published study population differed significantly from the population reported in the registry, particularly when it came to the description of comorbidities and the list of past or present therapies that were allowed in the trial. While it is acknowledged that it might be necessary to change the original eligibility criteria (e.g. adding an exclusion criterion because an unexpected side effect was detected in a subgroup of participants with a particular comorbidity), 7 these modifications should be clearly stated in the publication just like any other protocol amendment, as required by the Consolidated Standard of Reporting Trials (CONSORT) statement. 24 Limitations in space cannot be alleged to justify the omission of some eligibility criteria in publications, as these can be listed as supplemental material. A precise definition of the trial’s study population is essential for clinicals to ascertain if the results are applicable to patients seen in daily practice. In fact, a recent study evaluating the safety of MS drugs found out that patients not fulfilling certain inclusion criteria of their respective clinical trial were more likely to suffer from adverse events. 25 In our analysis, we came across the particular case of a clinical trial evaluating fingolimod in which the HbA1c threshold to exclude diabetic participants was set at 7.0% in the registry, but at 9.0% in the publication. This contradiction implies that neurologists cannot clearly tell if the study results apply for diabetic patients with intermediate to poor metabolic control. This is particularly striking in the case of fingolimod, since its well-known risk of macular oedema is especially high in diabetic individuals. The full knowledge of eligibility criteria is also of great interest to researchers conducting meta-analyses, since they allow to determine if cross-trials comparisons are feasible. Meta-analyses are often included in clinical practice guidelines and therefore have a strong influence on clinical decision-making.
Regarding primary outcome measures, we found out that nearly half of MS trials did not match the registry record, without a clear-cut predominance of inconsistencies favouring positive over negative results. However, since most inconsistencies were considered to have an undetermined effect, caution is needed before completely ruling out selective reporting bias. ClinicalTrials.gov has been previously used to demonstrate incomplete outcome reporting in trials coming from different fields of medicine13–16 and dentistry. 17 In these studies, the proportion of trials not matching the pre-specified primary outcome ranged from 18.5% to 50.0%, and there was not a clear evidence of selective reporting bias except for haematology trials. 15 It the particular case of MS, a study by Lemmens et al. 26 assessing clinical trials on DMTs that had been completed before December 2018 reported a lower proportion of trials with primary outcome inconsistencies than us (26.0% vs 48.2%). These differences could be due to the method used to retrieve and categorize information. First, in their analysis, they used the latest version of ClinicalTrials.gov’s record (which may have been updated after the trial’s end, potentially hindering the identification of post hoc changes), whereas we prioritized the version in force at the time of the first participant’s enrolment. Our approach aligns with Cochrane’s recommendations, 27 as it allowed us to thoroughly identify all subsequent protocol modifications made after the trial actually started. Note that we did not take into account protocol changes that were duly declared. Second, Lemmens et al. 26 did not consider differences in the completeness of outcome definitions to be inconsistencies, while we did so. We share the opinion of Zarin et al. 28 that vague outcome descriptions are relevant since ‘post hoc choices [. . .] could mask the fact that multiple comparisons were conducted, potentially invalidating the reported statistical analysis and allowing for cherry-picking of results’. Similarly to us, Lemmens et al. 26 did not find a significative predominance of inconsistencies favouring positive results.
With respect to the sample size estimation, data were missing or non-concordant in half of the publications, thus hindering if the number of study participants was enough to detect a statistically significant treatment effect (i.e. the trial statistical power). Undersized studies can lead to the erroneous assumption that the experimental drug is not more effective than placebo when in fact it is. But, even more alarmingly, the reader might wrongly conclude that the drug is safe just because the low sample size did not allow the detection of rare but otherwise serious side effects.
In our study, we focused on phase III and IV clinical trials, as we aimed to examine studies that directly influence marketing authorization and clinical decision-making rather than those related to early drug development (i.e. phase I and II). Considering the heterogeneity that could be introduced by analysing all trials together, we decided to assess whether the frequency of silent protocol modifications varied depending on study phase. Overall, we did not find remarkable differences between phase III and phase IV clinical trials in terms of protocol inconsistencies. Nor did we observe differences when analyzing the studies that specifically led to drug approval by the FDA or the EMA. However, the small sample included in these sub-analyses requires caution before fully assuming that no significant differences exist.
Consistent with other studies,15,26,29,30 we did not find an association between silent protocol changes and industry involvement. In addition, study registration status was neither associated with inconsistencies, that is to say, prospective registration itself did not eradicate incomplete outcome reporting nor other methodological discrepancies. Therefore, based on the findings of our study, we encourage editors and peer-reviewers not only to verify trial registration before accepting a publication, but also to actively review the accuracy of methodological data by cross-checking the manuscript and the registry record. If any silent protocol modifications were detected, authors and responsible parties should be asked for feasible explanations and encouraged to explicitly report those changes in the manuscript and discuss their influence on trial results. Ultimately, our findings call for a greater emphasis on the mandatory reporting of all protocol amendments and the promotion of a more standardized approach to documenting these changes.
Two limitations should be noted when interpreting the findings of our study. First, we only analysed trials registered in ClinicalTrials.gov. However, we consider it provides a representative sample given that it is the largest existing clinical trial registry by far. 11 Second, we excluded trial publications in languages other than English. This led to the exclusion of four clinical trials that had only been published in Russian. Nonetheless, the low proportion of excluded studies (3.4%) makes it unlikely that the results of our study have been biased because of language restrictions.
Conclusion
This study demonstrates that phase III and IV clinical trials examining MS drugs frequently have silent protocol changes which are only detectable by cross-checking journal publications and ClinicalTrials.gov. These methodological inconsistencies might distort the study interpretation and the judgement of the trial applicability. Therefore, efforts must be made to promote more transparency in MS clinical trials.
Supplemental Material
sj-docx-1-tan-10.1177_17562864251335247 – Supplemental material for Silent protocol modifications in multiple sclerosis clinical trials: a registry-based cross-sectional study
Supplemental material, sj-docx-1-tan-10.1177_17562864251335247 for Silent protocol modifications in multiple sclerosis clinical trials: a registry-based cross-sectional study by Alejandro Rivero-de-Aguilar, Mónica Pérez-Ríos, Joseph S. Ross, Marta Mascareñas-García, Alberto Ruano-Raviña, Marilina Puente-Hernandez and Leonor Varela-Lema in Therapeutic Advances in Neurological Disorders
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.