
Abstract
Background:
Alemtuzumab is administered intravenously (IV) for relapsing-remitting multiple sclerosis (RRMS), with limited studies of subcutaneous (SC) treatment.
Objectives:
We sought to evaluate the pharmacodynamics (PD), pharmacokinetics (PK), and safety profile of SC-administered alemtuzumab in people with progressive multiple sclerosis (PMS).
Design:
SCALA was a phase I, open-label, randomized, parallel-group study with two 12-month periods and a safety monitoring phase to 60 months.
Methods:
Of 29 screened participants, 24 were enrolled and randomized 2:1 to two 12 mg/day alemtuzumab treatments (60 and 36 mg total; SC:IV). Key inclusion criteria: ⩾18 years with a PMS diagnosis. Key exclusion criteria included RRMS diagnosis and prior treatment with anti-CD52 antibodies. Primary endpoint: CD3+ lymphocyte count. Secondary endpoints: PD and PK parameters.
Results:
Demographics were broadly similar for participants in the SC (16) and IV (8) arms; more participants with primary PMS received SC (44%) versus IV (25%) treatment. After the first course, the mean CD3+ cell count/µL was reduced at month 1 in both arms (SC: baseline (BL) 1326 to 48 vs IV: BL 1155 to 84). Lymphocyte counts partially repopulated by month 12, with mean CD3+ cell counts/µL of SC 599 versus IV 528. The mean lymphocyte counts/µL decreased again after the second course at month 13 in both arms (SC: 90 vs IV: 129), with partial repopulation by month 24. Alemtuzumab serum concentrations were lower following SC administration relative to IV, with 32% bioavailability. There were no adverse events leading to permanent treatment discontinuation or death.
Conclusion:
In SCALA, there were similar patterns of lymphocyte depletion and repopulation for participants receiving SC or IV alemtuzumab. In both arms, alemtuzumab had a manageable safety profile, with no emerging safety concerns. The general stabilization of neurological outcomes observed over 60 months underscores the potential long-term benefits of alemtuzumab treatment.
Trial registration:
Clinicaltrials.gov identifier: NCT02583594.
Keywords
Introduction
Multiple sclerosis (MS) is a chronic, immune-mediated disease that targets the central nervous system, characterized by neurological decline leading to disability. Most persons with MS initially have a relapsing-remitting course (RRMS), with a significant percentage eventually developing chronic progression, classified as secondary progressive MS (SPMS). Those with chronic progression from MS onset (approximately 10%) have primary progressive MS (PPMS). Many therapeutic options exist for RRMS; however, very limited treatment options are available for persons with progressive multiple sclerosis (PMS), which includes SPMS and PPMS.1,2 Disease-modifying therapies (DMTs) are critically required to alleviate the continuous increase in disability for persons with PMS.3,4
Alemtuzumab is a humanized anti-CD52 monoclonal antibody approved in over 70 countries worldwide. The recommended dosage regimen of alemtuzumab for RRMS is 12 mg/day administered by intravenous (IV) infusion for two initial treatment courses: 12 mg/day on 5 consecutive days (60 mg total dose) for the first course and 12 mg/day on 3 consecutive days (36 mg total dose) for the second course, administered 12 months after the first treatment. 5 In the phase III trials CARE-MS I 6 (NCT00530348) and CARE-MS II 7 (NCT00548405), IV alemtuzumab treatment resulted in selective depletion and a distinct pattern of repopulation of T and B lymphocytes in people with RRMS. Treatment with alemtuzumab demonstrated significantly greater improvements in efficacy outcomes compared with interferon beta-1a (IFNβ-1a), including decreased annual relapse rates in both trials and reduced disability accumulation in CARE-MS II. The main adverse events (AEs) reported in these trials were infusion-associated reactions (IARs), infections, thyroid disorders, and rarely, immune thrombocytopenia. Furthermore, a pilot study of 36 patients with SPMS treated with 100 mg alemtuzumab (20 mg/day for 5 days), with 7 patients re-treated after 2–4 years, reported a significantly reduced disability progression rate for at least 18 months compared with the year prior to treatment. A reduction in radiological markers of cerebral inflammation was also demonstrated, suggesting that one to two courses of alemtuzumab may suppress neuroinflammation. 8 These studies demonstrated that alemtuzumab treatment is effective in reducing relapse rates and in decreasing disability progression in MS.
Subcutaneous (SC) administration of alemtuzumab has been well tolerated in studies involving patients with chronic pure red cell aplasia, chronic lymphocytic leukemia, and cytoreduction prior to stem cell transplantation.9–11 More recent literature in oncology demonstrated improved tolerability with SC administration of alemtuzumab.12,13 In addition, a case report of two patients with highly active relapsing MS treated with SC alemtuzumab demonstrated significant improvement in disability and tolerability without infusion-associated AEs. 14 Furthermore, the advantages of SC administration over IV infusion, in terms of patient preference and reduction in treatment time, have been documented in several studies.15–18 Given the lack of clinical research on the pharmacodynamics (PD), pharmacokinetics (PK), and safety profile of SC-administered alemtuzumab in PMS, and limited information from patients with RRMS, 14 SCALA sought to evaluate these parameters of SC administration with alemtuzumab relative to IV infusion.
Methods
Participants
Of the 29 screened participants, 24 were eligible for study inclusion and enrolled in SCALA (Figure 1). Eligible participants were aged ⩾18 years with a diagnosis of PMS (including primary PMS and secondary PMS), based on the McDonald Criteria 2010 revision. 19 The exclusion criteria included a diagnosis of RRMS, prior treatment with alemtuzumab or other anti-CD52 antibodies, treatment with natalizumab in the 4 months prior to Study Visit 1, diagnosis of progressive multifocal leukoencephalopathy, treatment with an immunosuppressant or cytotoxic therapy other than steroids in the past 12 months, or treatment with the following DMTs: glatiramer acetate, IFNβ, or dimethyl fumarate in the past 4 weeks, fingolimod within the past 2 months, or teriflunomide within the past 12 months.

Trial flowchart. N/n = number of randomized patients in each treatment group. Percentages of items are calculated using the number of patients randomized as the denominator.
Study design
SCALA (NCT02583594) was a phase I, exploratory, single-center, open-label, randomized, parallel-group study conducted at Vall d’Hebron University Hospital in Barcelona, Spain, from November 30, 2015 to March 1, 2021. It comprised two 12-month study periods, and a safety monitoring phase extending to 60 months (Figure 2). A screening period of up to 28 days preceded the baseline (BL) visit in the first year (Period 1), followed by an initial treatment course (see treatment protocol) and 24 days of follow-up. There was an additional 11 months of monitoring for PD parameters, immunogenicity, and safety, with monthly visits. The second year (Period 2) included a pre-treatment visit preceding the second treatment course, 26 days of follow-up, and 11 months of monitoring as for Period 1. Years 3 through 5 (safety monitoring phase) spanned 36 months of monthly visits to monitor safety, disability, and neurological outcomes.
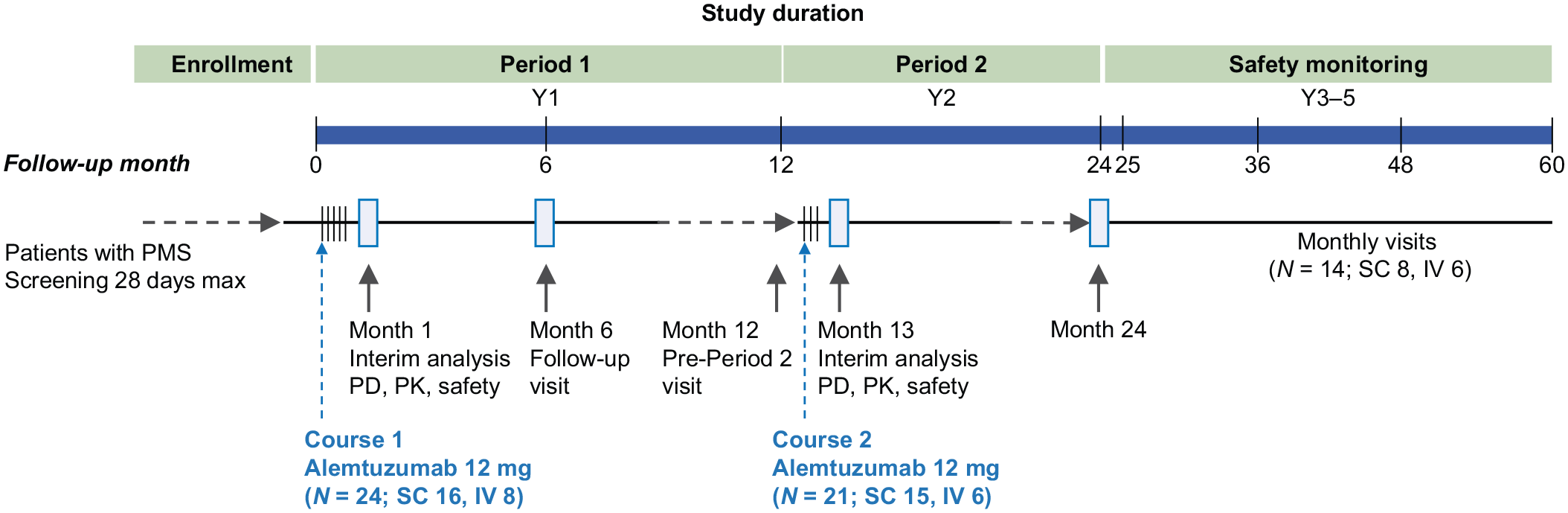
SCALA study design. SCALA comprised two 12-month study periods and a safety monitoring phase. Adults with primary or secondary PMS received two courses, SC or IV, of alemtuzumab 12 mg/day on 5 consecutive days at BL and on 3 consecutive days 12 months later. PD, PK, and safety assessments were undertaken in the interim analysis and as outlined in the section “Methods.” PD parameters were assessed at BL and months 1, 3, 6, 9, 12, 13, 15, 18, 21, and 24. CBC was assessed at BL, weeks 2, 3, and 4, and then monthly. Urinalysis and serum creatinine were assessed at BL and monthly. Biochemistry, including liver function tests, serum creatinine, and thyroid function tests, was analyzed at BL, month 1, month 13, and then quarterly. In the safety monitoring phase, AEs, CBC, serum creatinine, liver function tests, and urinalysis were analyzed monthly. At the month 24 visit, participants were asked to re-consent to continue receiving appropriate safety monitoring in a follow-up phase for 3 additional years.
Treatment protocol
Eligible participants were randomized in a 2:1 ratio (SC:IV) to receive two treatment courses of alemtuzumab administered either SC or IV. For each patient, the route of administration was identical for both treatment courses. The initial treatment course consisted of alemtuzumab administration of 12 mg/day SC or IV (1.2 mL/dose) on 5 consecutive days (60 mg total dose) from BL. Twelve months later, the second treatment course of 12 mg/day alemtuzumab was administered SC or IV on 3 consecutive days (36 mg total dose) (Figure 2).
Participants were pre-treated with 100 mg methylprednisolone orally the day before and immediately prior to alemtuzumab administration for both courses on the first 3 days of treatment. In addition, pre-treatment with antihistamines and/or antipyretics prior to alemtuzumab administration was undertaken at the investigator’s discretion. Oral prophylaxis for herpes infection was administered to all participants starting on the first day of alemtuzumab treatment and continuing for a minimum of 1 month.
PD parameters were assessed at BL and months 1, 3, 6, 9, 12, 13, 15, 18, 21, and 24. Serum samples for pharmacokinetic assessments were taken at BL, days 1–5 of the initial treatment course (2, 4, and 8 h after the first dose (day 1) and pre-dose (−1 h) of days 2–5), and during the post-treatment follow-up period (days 6–9, and days 11, 15, and 23). For the second treatment course, blood collection was taken on day −1, days 1–3 (2, 4, and 8 h after the first dose (day 1) and pre-dose (−1 h) of days 2 and 3), and during the post-treatment follow-up period (days 4–7). Complete blood count (CBC) was assessed at BL, weeks 2, 3, and 4, and then monthly. Urinalysis and serum creatinine were assessed at BL and monthly. Biochemistry, including liver function tests, serum creatinine, and thyroid function tests, was analyzed at BL, month 1, month 13, and then quarterly. In the safety monitoring phase, AEs, CBC, serum creatinine, liver function tests, and urinalysis were analyzed monthly. At the month 24 visit, participants were asked to re-consent to continue receiving appropriate safety monitoring in a follow-up phase for 3 additional years.
Outcomes
The primary endpoint was the CD3+ lymphocyte count after SC or IV administration. The secondary endpoints were PD parameters, including absolute counts of CD3+CD8+, CD3+CD4+, CD19+, and CD16+CD56+ lymphocyte subsets, total lymphocyte count, and helper/suppressor (CD4+/CD8+) ratio in the SC and IV arms. Lymphocyte phenotyping, including a standard, 6-color TBNK (T cells, B cells, and natural killer cells) panel (CD3+, CD4+, CD8+, CD19+, CD16+56+, total lymphocytes, and helper/suppressor (CD4+/CD8+) ratio), was performed. The TBNK panel and all subset panels (lymphocyte, T cell, B cell, dendritic cell/monocyte) were assessed at BL (pre-dose on day 1) and at quarterly visits for all participants, and additionally at months 1 and 13.
PK parameters, including bioavailability and Tmax, and safety were analyzed as secondary endpoints. Safety and tolerability were assessed, including AEs, serious AEs (SAEs), AEs of special interest (AESI), IARs, and injection site reactions (ISRs). The effects of alemtuzumab on disability and neurological functional status were examined by assessing the following exploratory variables over time: Expanded Disability Status Scale (EDSS) scores, Timed 25-Foot Walk Test (T25-FW), 9-hole peg test, Sloan contrast sensitivity chart test, symbol digit modalities test (SDMT), and the number of relapses requiring steroid treatment during the study.
Statistical analysis
The primary endpoint was summarized using descriptive statistics at each time point. Total lymphocytes were calculated by combining the count of CD3+, CD19+, and CD16+CD56+ cells. The secondary PD parameters for SC and IV administration were similarly summarized using descriptive statistics at each time point. Pharmacokinetic parameters were calculated using non-compartmental methods from serum alemtuzumab concentrations.
Results
Study population
Twenty-four participants were randomized 2:1 into alemtuzumab treatment arms (SC or IV). All 24 participants received course 1 (16 SC arm and 8 IV arm). Three participants (1 SC arm and 2 IV arm) discontinued the study due to their personal decision before starting course 2. A total of 21 participants (15 SC arm and 6 IV arm) completed course 2 and all were retained to month 24. At 60 months, a total of 14 (58%) participants completed the study, 8 (50%) in the SC arm and 6 (75%) in the IV arm. The most common reasons for discontinuation were patient decision and withdrawal from the study due to the addition of other DMTs, in compliance with the study exclusion criteria (Figure 1). Reasons for study discontinuation did not include AEs, poor compliance with protocol, or lack of follow-up.
Participant demographics and BL characteristics were broadly similar between the SC and IV arms (Table 1). There was a higher percentage of participants with PPMS in the SC arm (approximately 44%) relative to the IV arm (25%). The mean (standard deviation, SD) time since the first diagnosis of PMS was 6.00 (4.10) years in the SC arm and 6.38 (5.10) years in the IV arm. The mean (SD) number of relapses in the past 2 years prior to study entry was 0.25 (0.58) in the SC arm and 0.38 (0.74) in the IV arm and the median BL EDSS (range) was 5.75 (4.0–9.0) in the SC arm and 6.75 (4.0–7.5) in the IV arm. There were proportionally twice as many participants in the IV arm (25%) with previous MS medication in the past 12 months relative to participants in the SC arm (12.5%).
Patient characteristics at baseline.

This table depicts the characteristics of patients treated with 12 mg alemtuzumab SC or IV at baseline.
Patients were categorized as having PPMS using Cytel statistical software, where the number of “years since initial MS diagnosis” equals “years since first diagnosis for PMS.” Patients were otherwise classified as having SPMS.
BMI, body mass index; EDSS, Expanded Disability Status Scale; IV, intravenous; MS, multiple sclerosis; PMS, progressive MS; PPMS, primary progressive MS; SC, subcutaneous; SD, standard deviation; SPMS, secondary progressive MS.
Primary outcome
Following the first course of alemtuzumab treatment, the mean (SD) lymphocyte CD3+ cell count/µL was reduced from 1326 (443) at BL to 48 (48) at month 1 in the SC arm, and from 1155 (199) at BL to 84 (90) in the IV arm (Figure 3). Lymphocyte counts were partially repopulated by month 12, with mean (SD) CD3+ cell counts/µL of 599 (396) in the SC arm and 528 (331) in the IV arm. After the second alemtuzumab treatment course, lymphocyte counts decreased again in both treatment arms. The mean (SD) CD3+ cell count/µL at month 13 was 90 (71) in the SC arm and 129 (103) in the IV arm, with partial repopulation achieved by month 24.

Mean ± SD of the CD3+ lymphocyte cell count. The mean ± SD cell count of CD3+ lymphocytes (CD3+ cell count/µL) versus time (month) in patients treated with two courses of alemtuzumab. Patient numbers per visit are indicated below the graph.
Secondary outcomes
Pharmacodynamics
Depletion across other lymphocyte subpopulations, including CD3+CD8+, CD3+CD4+, CD19+, and CD16+CD56+ lymphocytes, total lymphocyte count, and CD4+/CD8+ ratio, was observed in the SC and IV arms by 1 month after alemtuzumab administration, with lymphocyte repopulation over time; a representative panel is shown in Figure 4. Lymphocyte values followed a similar course in both treatment arms, generally recovering toward BL levels during the first year after treatment. Following the second course of SC or IV alemtuzumab administration, lymphocyte depletion was observed by 1 month. The lymphocyte count repopulated again toward BL levels from month 13 to month 24. CD19+ lymphocytes recovered faster and above BL following each course of alemtuzumab.

Mean ± SD cell count of a representative panel of lymphocytes: (a) CD3+CD8+, (b) CD3+CD4+, (c) CD19+, and (d) total lymphocytes. Mean ± SD cell count (/µL) versus time (month) is shown in patients treated with two courses of alemtuzumab. Patient numbers per visit are indicated below the graphs.
Pharmacokinetics
Alemtuzumab serum concentrations increased after consecutive daily SC or IV administration, with a higher Cmax after five consecutive doses compared with three consecutive doses. Serum concentrations were lower following SC administration relative to IV, with a bioavailability of 32% versus IV. Serum levels decreased over time, with IV serum concentrations slowly declining until both treatments had similar concentrations by 336 h post-dose. Alemtuzumab was absorbed with a median Tmax of approximately 4 or 6 days following IV or SC treatment, respectively. Following IV treatment, alemtuzumab concentrations were measurable during the entire sampling period (22 days) and declined slowly with a t1/2z of 6.79 days (N = 1). Following SC treatment, alemtuzumab was absorbed slowly into the systemic circulation and eliminated slowly (t1/2z of 9.21 days) with measurable serum concentrations up to the last sampling time of 22 days.
Safety
Safety and tolerability profiles in both treatment arms were manageable, with no emerging safety concerns (Table 2). There were no AEs leading to permanent treatment discontinuation or death.
Safety data.
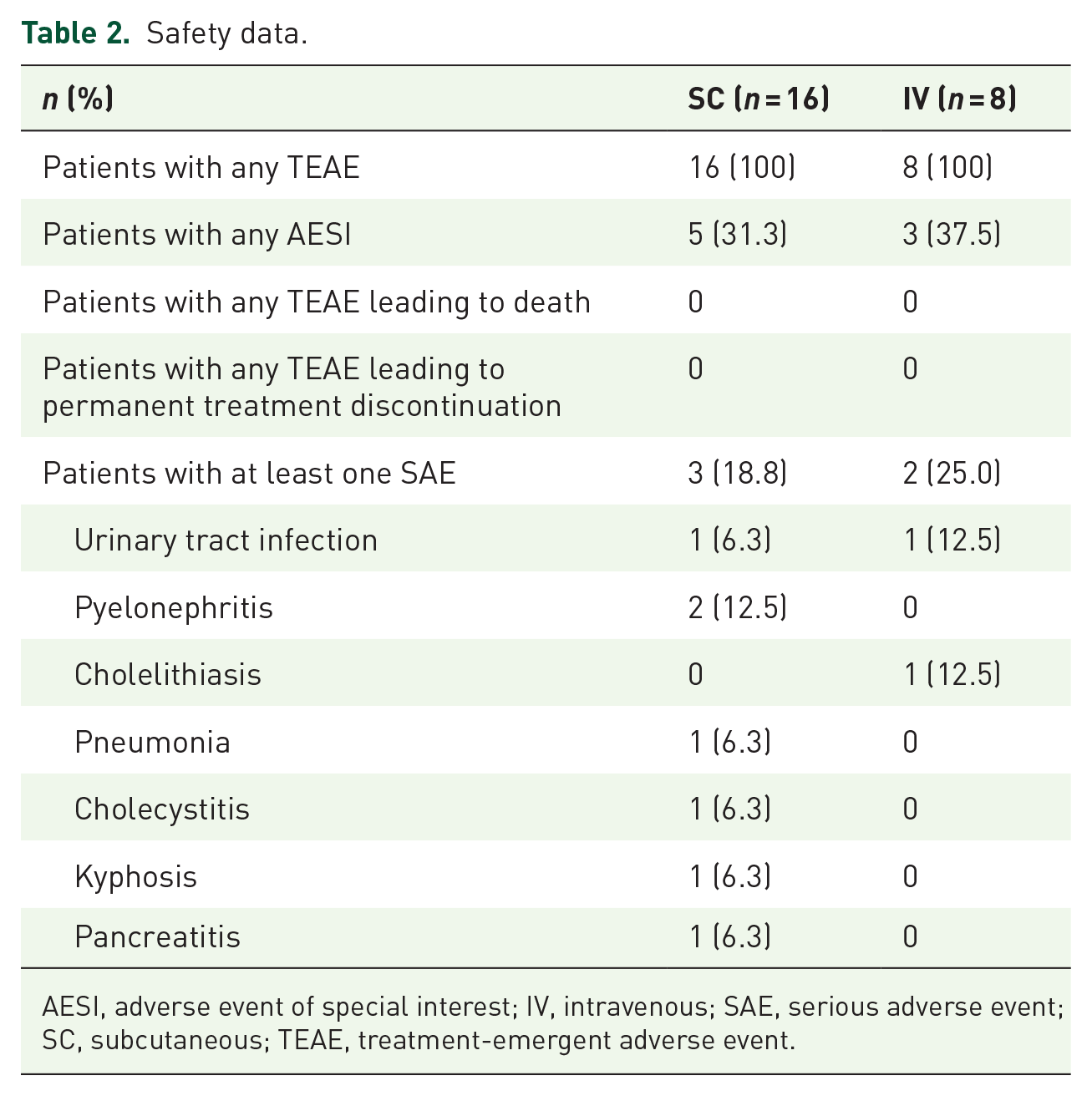
AESI, adverse event of special interest; IV, intravenous; SAE, serious adverse event; SC, subcutaneous; TEAE, treatment-emergent adverse event.
The percentage of treatment-emergent AEs (TEAEs) was similar in both SC and IV arms, and there were more severe TEAEs in the SC group compared with IV infusion. The most commonly reported TEAEs following SC administration (n = 16) were injection site erythema (15), pyrexia (15), headache (11), urinary tract infection (UTI; 9), respiratory tract infection (8), and upper respiratory tract infection (8). The most commonly reported TEAEs following IV administration (n = 8) were pyrexia (8), headache (7), and upper respiratory tract infection (5). Additional TEAEs in the SC arm included hypothyroidism (1) and hyperthyroidism (2). In the IV arm, hypothyroidism (1), sinus tachycardia (1), and drug hypersensitivity (1) were observed, with no reports of immune thrombocytopenia. Three participants reported severe (Grade 3) TEAEs following SC administration; no Grade 4 or 5 TEAEs were reported. No severe (Grade ⩾3) TEAEs were reported in the IV arm.
Seven AESIs were reported in five participants (31.3%) in the SC arm: antiphospholipid antibodies (1), hypothyroidism (1), hyperthyroidism (2), pyelonephritis (2), and pneumonia (1), of which three AESIs were serious (pyelonephritis (2), pneumonia (1)). The antiphospholipid antibodies were identified during the diagnostic workup of skin lesions. No major thrombotic event occurred. Three AESIs occurred in three participants (37.5%) in the IV arm: hypothyroidism (1), pregnancy (1), and UTI (1), of which one AESI was serious (UTI).
Seven SAEs occurred in three (18.8%) participants following SC treatment: pyelonephritis (n = 2), UTI, and pneumonia were considered related to alemtuzumab. One of these participants required surgery for a previous kyphosis, which was complicated by cholecystitis and pancreatitis during hospitalization; these SAEs were not considered related to alemtuzumab. This participant also reported pyelonephritis, which was considered related to alemtuzumab. In the IV arm, SAEs occurred in two participants (25%): UTI and cholelithiasis; neither was considered related to alemtuzumab.
IARs were defined as any AE occurring during alemtuzumab infusion or within 2 h post-infusion. Those reported in the IV group (n = 8) included pyrexia (8), headache (5), temperature regulation disorder (3), neurological decompensation, sinus tachycardia, bronchospasm, hypoxia, nausea, pruritus, rash erythematous, and injection site pruritus (1 each). ISRs reported in the SC group (n = 16) included mild erythema (9), moderate erythema (5), and erosion on the injection site (1), with no reports of severe or potentially life-threatening erythemas.
Immunogenicity
In the SC arm, 4/16 (25.0%) participants reported negative antidrug antibody (ADA) analyses, and 12/16 (75.0%) participants reported positive ADA during the study. In the IV arm, all 8 participants reported positive ADA. Of all IV participants reporting positive ADA, 2/8 (25.0%) participants reported transient treatment-induced ADA, whereas the remaining participants reported persistent treatment-induced ADA.
Exploratory outcomes
MS activity and disability
Two participants had relapses following SC treatment: one participant had one relapse in the period between BL and month 12, and another participant had two relapses between month 12 and month 24. No relapses were reported in the IV arm during the study. No relapses were reported from month 24 to the end of study (EOS) in either treatment arm.
The median EDSS score for the SC arm increased from 5.75 at BL to 7.00 at EOS (Figure 5). The median EDSS score for the IV arm remained stable at 6.75, at both BL and EOS, despite a median change of 0.25 relative to BL during the study.

EDSS scores over time. Box plot of EDSS scores from BL to EOS in (a) SC and (b) IV treatment arms. The bottom and top edges of the box represent the interquartile range. The line inside the box represents the median value, which is also indicated by the number. The whiskers are drawn from the box to the minimum and maximum scores. Patient numbers per visit are indicated below the graphs.
Participants were further assessed using disability and neurological tests as an exploratory endpoint (Supplemental Figure S1). The median time (seconds) to complete the T25-FW was stable in both the SC and IV arms (SC: 14.20 BL vs 13.10 month 24; IV: 10.00 BL vs 9.70 month 24). There was an increase relative to BL in the median time (seconds) to complete the 9-hole peg test in the SC arm (30.20 BL vs 34.30 month 24) and a decrease relative to BL in the IV arm (34.63 BL vs 30.50 month 24). For the Sloan contrast sensitivity chart test, there was a decrease in the total median number of correct binocular eye tests in the SC arm (48.00 BL vs 43.00 month 24), and a similar median number of correct tests relative to BL in the IV arm (44.50 BL vs 44.00 month 24). There was an increase relative to BL in the total median number of correct responses for the SDMT (SC: 31.50 BL vs 36.00 month 24; IV: 35.00 BL vs 38.00 month 24). Overall, minor changes were observed during 60 months for the SC arm of the Sloan contrast sensitivity chart test, and for both treatment arms of the 9-hole peg test and the SDMT, with results suggesting a general stabilization relative to BL.
Discussion
In this phase I study, we compared SC versus IV administration of alemtuzumab using a participant population with PMS and severe disability (Table 1). SC and IV treatments resulted in similar patterns of lymphocyte depletion and repopulation following alemtuzumab administration. This occurred with CD3+ lymphocytes, total lymphocytes, and additional lymphocyte subsets tested, including CD19+ lymphocytes which showed faster recovery and repopulation above BL after alemtuzumab treatment (Figures 3 and 4). The first case report of alemtuzumab SC treatment in two patients with RRMS also demonstrated depletion in total lymphocyte count, which occurred within days following a sole treatment course (12 mg/day SC for 5 days), with a subsequent increase of total lymphocytes in 2–3 months. 14 In SCALA, which compared alemtuzumab SC and IV treatments for patients with PMS using two treatment courses, depletion of total lymphocytes occurred within 1 month, and partial repopulation occurred by 12 months. The therapeutic effect of alemtuzumab may involve immunomodulation through depletion and repopulation of lymphocytes. 20 The similar pattern of lymphocyte depletion and repopulation for both routes of alemtuzumab administration in SCALA suggests that SC treatment may result in similar therapeutic effects to IV administration.
Pharmacokinetic analyses in SCALA indicated that alemtuzumab serum concentrations increased similarly following SC or IV administration, with a higher Cmax after the first treatment course. This is consistent with previous trials of alemtuzumab IV infusion. 20 Drug absorption occurred with a median Tmax of 6 days for SC treatment compared with 4 days for IV. It is uncertain whether the slower absorption in the SC arm and decreased bioavailability following administration impacts therapeutic efficacy.
Previous phase II and phase III trials in RRMS demonstrated that alemtuzumab had greater efficacy relative to IFNβ-1a, including a decreased relapse risk, improvement or stabilization in disability and EDSS scores, and reduced brain volume loss.21,22 The maximum EDSS score was 5.0 for participants in these trials. In SCALA, BL EDSS scores ranged from 4.0 to 9.0, with no relevant worsening in the IV arm over 60 months in this small population. A sustained effect of alemtuzumab has been reported in other extension studies, such as the CARE-MS extension trial (CAMMS03409), 23 and the TOPAZ extension study. 24 These long-term benefits of alemtuzumab may be due to a neuroprotective effect, as suggested by in vitro studies indicating that lymphocyte repopulation following alemtuzumab treatment may be involved in brain repair. 25 A “therapeutic window” for the lasting clinical benefits of alemtuzumab from this potential neuroprotective effect was proposed based on an earlier study of participants with SPMS. 26 There was an absence of new magnetic resonance imaging (MRI) lesions 7 years after treatment with alemtuzumab, yet participants still demonstrated sustained accumulation of disability and increased cerebral atrophy. By contrast, participants with RRMS treated prior to the progressive phase reported reduced disability up to 36 months after alemtuzumab treatment. The authors hypothesized a time frame early in the course of MS, when neuroprotection from inflammation and demyelination by immunotherapy may prevent long-term axonal loss. 26 Thus, treating participants with alemtuzumab earlier in the disease course, during the suggested “therapeutic window,” resulted in improved long-term clinical outcomes. Participants in SCALA had advanced progressive MS. This may explain the increase in EDSS scores at EOS relative to BL in the SC arm (Figure 5(a)). The general stabilization relative to BL observed in the SCALA population suggests that people with high EDSS may benefit from alemtuzumab treatment. However, participants are limited by the late stage of the disease and may not benefit from the suggested “therapeutic window.” In addition, participants with lower versus higher EDSS may incur greater benefits from the hypothesized neuroprotection in response to alemtuzumab treatment.
Alemtuzumab was well tolerated in both treatment arms of this study relative to previous trials, considering the advanced disease state of this population. An increased incidence of TEAEs was reported after SC administration compared with IV infusion, with no AEs leading to permanent treatment discontinuation or death (Table 2). Thyroid events have been reported in alemtuzumab studies.6,7,27 In the CARE-MS I and CARE-MS II trials, 18% and 16% of participants developed thyroid AEs, respectively.6,7 In this study, thyroid AEs were consistent with previous findings, with 18.75% of participants developing thyroid disorders in the SC arm (hypothyroidism (1) and hyperthyroidism (2)), and a lower percentage of participants (12.5%) with thyroid disorders in the IV arm (hypothyroidism (1)).
During infusion, IARs may occur due to allergic hypersensitivity or cytokine release.28,29 The IARs seen in SCALA were generally consistent with those reported in other trials,6,7,30,31 even though higher doses of methylprednisolone were applied (1000 mg) in the CARE-MS I and CARE-MS II studies. In addition, the population characteristics in SCALA (older and with greater disabilities) may render participants more susceptible to IARs. One case (1/8) of sinus tachycardia was reported as an IAR in this study; CARE-MS I and CARE-MS II reported 29/376 and 21/435 cases, respectively. 32 Infections reported in SCALA, including respiratory infections and UTIs, were also seen in the CARE-MS I and CARE-MS II trials.6,7 The incidence of AEs, including thyroid disorders and infections following alemtuzumab treatment, was found to decrease over time in extension studies up to 9–12 years.24,31
This study was limited by the small size of the population, which made direct comparisons between treatment arms more difficult. The sample size was not based on statistical power due to the exploratory nature of the trial, which was conducted at a single site, thereby restricting the study population to a specific demographic group. There was a difference in the ratio of patients with PPMS to SPMS in the SC and IV treatment arms (Table 1), with the SC population being older, which may account for observed differences in disability outcomes (Figure 5). Although 93.8% of participants in the SC arm completed Period 2 of the main study, only 50% of participants in the SC arm completed the entire study (Figure 1). Furthermore, the lack of MRI data did not allow correlation of each alemtuzumab administration route with disease activity or progression. Further studies are warranted to elucidate the benefit/risk profile of alemtuzumab SC versus IV administration in persons with MS.
Conclusion
In the SCALA trial, there was no overall difference observed in lymphocyte depletion and repopulation between the SC and IV alemtuzumab treatment arms. Alemtuzumab had a manageable safety profile in both treatment arms, with no emerging safety concerns. Over the 5-year study, stabilization relative to BL was generally observed in the number of relapses, disability, and neurological function tests in both treatment arms but was more pronounced in the IV arm. These safety and efficacy outcomes are clinically relevant given the progressive disease and advanced disability of the SCALA population, and they highlight the long-term benefit/risk profile of alemtuzumab treatment.
Supplemental Material
sj-docx-1-tan-10.1177_17562864241291655 – Supplemental material for SCALA: a randomized phase I trial comparing subcutaneous and intravenous alemtuzumab in patients with progressive multiple sclerosis
Supplemental material, sj-docx-1-tan-10.1177_17562864241291655 for SCALA: a randomized phase I trial comparing subcutaneous and intravenous alemtuzumab in patients with progressive multiple sclerosis by Xavier Montalban, Breogan Rodriguez-Acevedo, Carlos Nos, Mireia Resina, Mireia Forner, Yanzhen Wu and Magdalena Chirieac in Therapeutic Advances in Neurological Disorders
Footnotes
Acknowledgements
The authors and Sanofi thank the participants and their caregivers for their participation in the trial. Writing assistance, including assistance drafting and editing the manuscript text, figures, and tables, as directed by the authors; data checking and incorporation of comments from reviewers; and assisting with the submission process, was provided by Rania Kairouz-Wahbe, PhD, and Renee E. Granger, PhD, of Envision Pharma Group.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.