
Abstract
Background:
Generalized myasthenia gravis (gMG) is a chronic, unpredictable disease associated with high treatment and disease burdens, with a need for more effective and well-tolerated treatments.
Objectives:
To evaluate the long-term safety, tolerability, and efficacy of zilucoplan in a mild-to-severe, acetylcholine receptor autoantibody-positive (AChR+) gMG population.
Design:
Ongoing, multicenter, phase III open-label extension (OLE) study.
Methods:
Eligible patients had completed a qualifying randomized, placebo-controlled phase II or phase III zilucoplan study and received daily, self-administered subcutaneous 0.3 mg/kg zilucoplan. The primary endpoint was incidence of treatment-emergent adverse events (TEAEs). Secondary efficacy endpoints included change from baseline in Myasthenia Gravis Activities of Daily Living (MG-ADL) score.
Results:
In total, 200 patients enrolled. At the cut-off date (8 September 2022), median (range) exposure to zilucoplan in RAISE-XT was 1.2 (0.11–4.45) years. Mean age at OLE baseline was 53.3 years. A total of 188 (94%) patients experienced a TEAE, with the most common being MG worsening (n = 52, 26%) and COVID-19 (n = 49, 25%). In patients who received zilucoplan 0.3 mg/kg in the parent study, further improvements in MG-ADL score continued through to Week 24 (least squares mean change [95% confidence interval] from double-blind baseline −6.06 [−7.09, −5.03]) and were sustained through to Week 60 (−6.04 [−7.21, −4.87]). In patients who switched from placebo in the parent study, rapid improvements in MG-ADL score were observed at the first week after switching to zilucoplan; further improvements were observed at Week 24, 12 weeks after switching (−6.46 [−8.19, −4.72]), and were sustained through to Week 60 (−6.51 [−8.37, −4.65]). Consistent results were observed in other efficacy endpoints.
Conclusion:
Zilucoplan demonstrated a favorable long-term safety profile, good tolerability, and sustained efficacy through to Week 60 with consistent benefits in a broad AChR+ gMG population. Additional long-term data will be available in future analyses.
Trial registration:
ClinicalTrials.gov identifier: NCT04225871 (https://clinicaltrials.gov/ct2/show/NCT04225871)
Introduction
Myasthenia gravis (MG) is a chronic autoimmune disease, characterized by fluctuating muscle weakness and exertional fatigue, that affects between 100 and 350 patients per million people globally.1–3 Limitations with some current treatments include a long latency before therapeutic effect and risk of systemic adverse events, leaving up to 50% of patients with inadequately controlled disease, despite treatment.4,5 Patients can continue to experience unpredictable exacerbations and myasthenic crises, especially in the first year after diagnosis, highlighting the need for additional treatments that offer rapid onset of action and long-term, consistent, and sustained symptom improvements. 6
Long-term data are emerging for targeted therapies for the treatment of generalized MG (gMG), which includes the complement component 5 (C5) inhibitors eculizumab 7 and ravulizumab.8,9 More recently, phase III data for zilucoplan, a small (15-amino-acid) macrocyclic peptide C5 inhibitor with a dual mechanism of action, self-administered as a once-daily subcutaneous injection, have demonstrated both the efficacy of C5 inhibition and tolerability of daily subcutaneous administration in patients with acetylcholine receptor autoantibody-positive (AChR+) gMG, thus supporting the potential of zilucoplan as a next-generation C5 inhibitor. 10 In the pivotal 12-week, phase III, randomized, double-blind (DB), placebo-controlled RAISE study (NCT04115293), treatment with zilucoplan resulted in rapid, consistent, sustained, statistically significant, and clinically meaningful improvements from baseline, compared with placebo, in several well-established and MG-specific patient- and clinician-reported outcomes in patients with AChR+ gMG. Zilucoplan was also well tolerated with a favorable safety profile. 10
Zilucoplan targets the complement pathway by binding to C5 with high specificity and affinity to prevent C5 cleavage to C5a and C5b. In addition, zilucoplan binds to the C5b domain of C5 to sterically hinder binding of C5b to C6, which prevents the subsequent assembly and activity of the membrane attack complex, should any C5b be formed.2,11 This dual mechanism of action differs from that of eculizumab and ravulizumab, which only prevent C5 cleavage to C5a and C5b.12,13 Further, the binding of zilucoplan is not affected by the C5 p.Arg885His polymorphism, which is present in some East Asian patients, and is associated with poor response to eculizumab. 14 In vitro data have found zilucoplan to inhibit complement activation in patients with this polymorphism.15,16
RAISE-XT (NCT04225871) is an ongoing open-label extension (OLE) study of zilucoplan in adult patients with gMG. The primary objective is to evaluate the long-term safety and tolerability of zilucoplan. In this interim analysis, we aimed to assess long-term safety, tolerability, efficacy, and patient satisfaction with self-injection.
Methods
Study design
RAISE-XT is a multicenter OLE study of zilucoplan in patients with AChR+ gMG who have previously completed either the phase II 11 or phase III 10 studies of zilucoplan (Supplemental Figure 1). Patients assigned to the placebo arm of the phase II study were initially re-randomized to receive either 0.1 or 0.3 mg/kg zilucoplan for an extension period of the phase II study. Following a protocol amendment to the phase II study in April 2019, all patients who continued into the phase II extension received 0.3 mg/kg zilucoplan, as the dose selected for the phase III study. Patients who had already started with 0.1 mg/kg switched to 0.3 mg/kg. Thus, on entry into RAISE-XT, patients were entered in one of four treatment groups, as shown in Figure 1 and Supplemental Figure 1. For the safety analysis, patients were also assessed in one group, regardless of treatment or dose in the parent study (all ZLP).
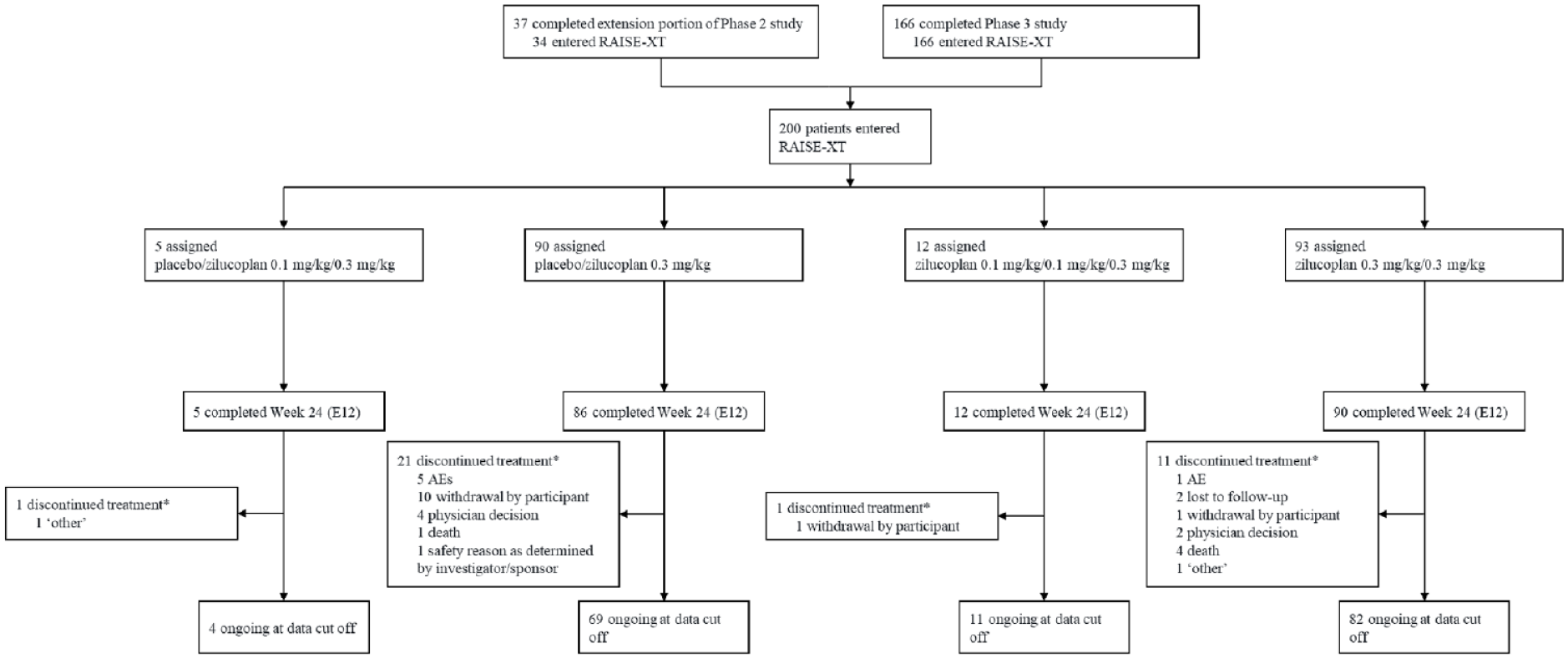
Patient disposition.
The data cut-off date for this prespecified interim analysis was 8 September 2022. An institutional review board or independent ethics committee for each participating site approved the protocol (Supplemental Material). This trial is registered with ClinicalTrials.gov (NCT04225871).
Patients
Inclusion and exclusion criteria for the phase II and phase III studies were similar and have been reported in detail elsewhere.10,11 Briefly, patients were adults aged ⩾18 years, diagnosed with mild-to-severe AChR+ gMG [Myasthenia Gravis Foundation of America (MGFA) Disease Class II–IV at screening] and had a Quantitative Myasthenia Gravis (QMG) score of ⩾12. One notable difference in inclusion criteria between the two qualifying studies is that the phase II study did not require a minimum Myasthenia Gravis Activities of Daily Living (MG-ADL) score, unlike the phase III study, in which patients were required to have an MG-ADL score of ⩾6 at screening and baseline. Patients were also required to have a quadrivalent and, where available, serotype B meningococcal vaccine. A booster vaccination should also have been administered as clinically indicated, according to local standard of care, for patients who were previously vaccinated against Neisseria meningitidis.
Patients were excluded if they were pregnant, planning to become pregnant, or nursing; were concurrently participating in another clinical trial involving an experimental intervention, with the exception of a prior zilucoplan trial, observational studies, or registry studies; had commenced any disallowed medication per the exclusion criteria from the qualifying zilucoplan study or altered the dose of any other concomitant medication, unless medically indicated; had any new or worsening medical condition since entry into the qualifying zilucoplan study; or had developed hypersensitivity to zilucoplan, any of its excipients or placebo.
For eligible patients opting to enroll from the phase III study, the last visit served as their first visit of the OLE (Day E1), which included review of eligibility to continue. Eligible patients transitioning from the phase II extension period could join the RAISE-XT OLE at their next study visit, without needing to repeat previously completed visits. All patients provided written informed consent and could withdraw consent at any time.
Intervention
Subcutaneous doses of zilucoplan were self-administered daily at home at approximately the same time each day. Doses were supplied as a sterile, preservative-free, aqueous solution in prefilled 1 mL glass syringes with a 29-gauge, ½-inch, staked needle placed within a self-administration device (BD Ultrasafe Plus™; BD Medical, NJ, USA). Patients could receive intravenous immunoglobulin (IVIg) or plasma exchange (PLEX) treatment as rescue therapy concomitantly with zilucoplan if, per the investigator’s judgment, escalation of gMG therapy became necessary due to deterioration of their clinical status or risk of MG crisis. ‘MG worsening’ could be reported as a treatment-emergent adverse event (TEAE) per the investigator’s judgment, and this was not limited to patients who received rescue therapy.
Outcome measures
During the first 12 weeks of the OLE, safety, tolerability, and efficacy were assessed at Extension Weeks E1 (Week 13; 1 week after 12-week DB period), E2 (Week 14), E4 (Week 16), E8 (Week 20), and E12 (Week 24). From Week E12 (Week 24), monthly visits were required for reporting of any changes to concomitant medication and adverse events, and quarterly visits were required from Week E24 (Week 36) for study assessments, including efficacy.
The primary outcome was incidence of TEAEs. TEAEs were defined as an adverse event starting on or after the time of the first administration of zilucoplan in the OLE and up to and including 40 days after the final dose (or final contact, whichever occurred first). Safety was additionally assessed by vital signs, physical examinations, electrocardiograms, clinical laboratory tests, antibody titers, and the Columbia-Suicide Severity Rating Scale.
Secondary outcomes were change from baseline to Week 24 in MG-ADL, QMG, Myasthenia Gravis Composite (MGC), and the revised 15-item Myasthenia Gravis Quality of Life (MG-QoL 15r) scores, and use of rescue therapy. Change from baseline to Week 60 and change from Week 12 to Week 60 were assessed. Exploratory efficacy endpoints included achievement of Minimal Manifestation Status per MGFA-post intervention status (MGFA-PIS; minimal manifestation is defined as no symptoms of functional limitations from MG, but with some weakness on examination of some muscles) without rescue therapy; Work Productivity and Activity Impairment: Specific Health Problem (WPAI:SHP), EQ-5D-5L, and Quality of Life in Neurological Disorders (Neuro-QoL) Short Form fatigue scale (raw scores); proportion of patients achieving a ⩾3-point reduction in MG-ADL score (MG-ADL responder), and ⩾5-point reduction in QMG score from baseline (QMG responder), without rescue therapy; and proportion of patients achieving minimal symptom expression (MSE; defined as MG-ADL score of 0 or 1 without rescue therapy). MGFA-PIS, WPAI:SHP, EQ-5D-5L, and Neuro-QoL Short Form fatigue scale were not included in the phase II study.
The self-injection experience was assessed by the Self-Injection Assessment Questionnaire (SIAQ; Version 2.0 POST module) in a subgroup of patients enrolled from sites in the United States only, due to a country-specific protocol amendment that added SIAQ as an additional exploratory endpoint. Patients completed the questionnaire on two occasions approximately 2 weeks apart, directly after self-injection. Scores for each of the six SIAQ domains range from 0 (worst experience) to 10 (best experience). 17 Pharmacodynamic (PD) outcomes included assessment of complement activity using a sheep red blood cell lysis assay. 18 Antidrug antibodies (ADAs) were also assessed using blood samples taken at OLE baseline (Week 12), Week 16, Week 24, and at quarterly visits thereafter.
Statistical methods
While no formal power calculation was done to determine sample size for the OLE, it was assumed that approximately 200 patients would enroll in RAISE-XT from the qualifying parent studies and remain in the study for an average of 2 years, thus providing approximately 400 patient-years of exposure. Sample size calculations for the qualifying parent studies are described elsewhere.10,11 Safety assessments (primary analysis) were performed on the Safety Set, which included all patients who received at least one dose of zilucoplan in RAISE-XT. Efficacy analyses were performed on the modified intent-to-treat population, which included all enrolled patients in RAISE-XT who received at least one dose of zilucoplan and had at least one post-dosing MG-ADL score. Two baselines were used: DB study baseline (Week 0) and OLE baseline (Week 12; or the last available assessment before the first administration of zilucoplan in the OLE).
Change from baseline up to Week 60 in MG-ADL, QMG, MGC, and MG-QoL 15r scores was estimated using a linear mixed model repeated measures (MMRM) analysis of covariance, with baseline MG-ADL score, baseline QMG score, baseline score (for MGC and MG-QoL 15r only), geographical region, qualifying study factor, visit and baseline score × visit (interaction term) as fixed effects, and participant as a random effect using an unstructured correlation structure. Separate models were fitted for each group (PBO/ZLP 0.3 and ZLP 0.3/ZLP 0.3). Least squares (LS) means and 95% confidence intervals were provided at each visit, with LS mean difference to compare Week 24 to Week 12 (at the end of the DB study). All safety and efficacy data were summarized by descriptive statistics. For continuous variables, mean and standard deviation are presented. For categorical variables, the number and percentage of patients in each category are presented. All observed data were used regardless of any intercurrent event; no data were censored.
Results
Participants and baseline demographics
In total, 200 patients enrolled in RAISE-XT and were included in the safety and efficacy analyses (Figure 1). Thirty-four (17%) and 166 (83%) patients were enrolled from the phase II and phase III studies, respectively, including all patients who completed RAISE, and, at the time of data cut-off, most patients (83%) were continuing to receive zilucoplan in the study. Median (range) exposure to zilucoplan in the OLE was 1.2 (0.11–4.45) years, leading to a total duration of exposure of 321.4 patient-years. The impact of COVID-19 on planned visits and assessments was minimal during the study.
A broad gMG population with mild-to-severe gMG as per the MGFA disease classification was enrolled (Table 1, Supplemental Table 1). Mean MG-ADL scores at DB baseline for patients who enrolled from the phase II study (PBO/ZLP 0.1/ZLP 0.3; n = 5 and ZLP 0.1/ZLP 0.1/ZLP 0.3; n = 12 groups) were slightly lower (8.4 and 7.2, respectively) than for the treatment groups including patients from the phase III study [10.7 (PBO/ZLP 0.3; n = 90) and 9.9 (ZLP 0.3/ZLP 0.3; n = 93)], as expected, since the phase II study did not require a minimum MG-ADL score.
Patient demographics and characteristics at RAISE-XT baseline.

ITT population. Baseline was defined as the last available assessment before first administration in the open-label period.
Refractory status was not recorded for patients in the phase II study. The N for ‘placebo/zilucoplan 0.3 mg/kg’, ‘zilucoplan 0.3 mg/kg/0.3 mg/kg’, and ‘all zilucoplan’ groups were 84, 82, and 166 patients, respectively.
Baseline medications include any medications that started prior to dosing in the OLE and continued after (classified as prior and concomitant medications).
gMG, generalized myasthenia gravis; IST, immunosuppressive therapy; ITT, intention-to-treat; MG-ADL, Myasthenia Gravis Activities of Daily Living; MGFA, Myasthenia Gravis Foundation of America; NA, not applicable; OLE, open-label extension; QMG, Quantitative Myasthenia Gravis; SD, standard deviation.
Safety analyses
Overall, 188 (94%) patients experienced TEAEs, and 64 (32%) patients experienced serious TEAEs (Table 2) during the OLE. The most common TEAEs were MG worsening [n = 52, 26%; of whom 22 (42%) received rescue therapy], COVID-19 (n = 49, 25%), headache (n = 35, 18%), diarrhea (n = 30, 15%), and nasopharyngitis (n = 30, 15%). The most common serious TEAEs were MG worsening (n = 15, 8%) and COVID-19 pneumonia (n = 4, 2%). Treatment-related serious TEAEs were reported in two (1%) patients overall: one event of esophagitis (ZLP 0.3/ZLP 0.3 group); and one event of injection site infection (occurring on the right inner thigh, which is not a recommended injection site 10 ; ZLP 0.3/ZLP 0.3 group). The most common treatment-related TEAE was injection site bruising, occurring in 12 (6%) patients.
Overview of treatment-emergent adverse events.

Safety set. Most common TEAEs occurring in ⩾15% of patients overall and most common serious TEAEs occurring in ⩾2% patients are reported only. Preferred terms listed as per MedDRA Version 24.0 descriptions.
Includes all AEs of death.
AE, adverse event; CI, confidence interval; COVID-19, coronavirus disease 2019; TEAE, treatment-emergent adverse event.
As of the clinical cut-off date, the majority of TEAEs were mild (50 patients, 25%) or moderate (81 patients, 41%). Seventeen (9%) patients had a TEAE resulting in permanent withdrawal from treatment or an AE of death, of whom five (3%) patients discontinued due to MG worsening. Two (1%) patients had treatment-related injection site reactions resulting in permanent withdrawal. One patient in the PBO/ZLP 0.3 group discontinued due to a nonserious treatment-related lipase increase that had resolved by the cut-off date. TEAEs resulting in death occurred in four (2%) patients overall, including cardiac arrest in two patients with major cardiovascular risk factors and one accidental head injury in the ZLP 0.3/ZLP 0.3 group, and one death from an unknown cause in a patient in the PBO/ZLP 0.3 group, who had major cardiovascular risk factors and severe pneumonia that had started 2 days earlier. No deaths were considered treatment related.
Efficacy analyses
Efficacy data are reported for the PBO/ZLP 0.3 (n = 90) and ZLP 0.3/ZLP 0.3 (n = 93) groups only due to low patient numbers in the PBO/ZLP 0.1/ZLP 0.3 (n = 5) and ZLP 0.1/ZLP 0.1/ZLP 0.3 (n = 12) groups, and in anticipation of a possible influence on efficacy after receiving 0.1 mg/kg zilucoplan in the OLE period of the phase II study before the protocol amendment.
In the ZLP 0.3/ZLP 0.3 group, mean MG-ADL, QMG, MGC, MG-QoL 15r, and Neuro-QoL Short Form fatigue scores improved from DB baseline to Week 12, continued to improve further through to Week 24, and were sustained through to Week 60 [Figure 2(a)–(e)]. In the PBO/ZLP 0.3 group, rapid improvements were observed at the first week after switching to zilucoplan 0.3 mg/kg (Week E1/Week 13) in MG-ADL, QMG, MGC, MG-QoL 15r, and Neuro-QoL Short Form fatigue score [Figure 2(a)–(e)]. Further improvements were observed through to Week 24, after 12 weeks of active treatment, and sustained through to Week 60, and were clinically meaningful for MG-ADL, QMG, and MGC as per the published clinical meaningfulness thresholds.19–21 At the time of the study, no threshold for clinical meaningfulness for changes in the MG-QoL 15r score had been established. Radar plots presenting mean MG-ADL, QMG, MGC, and MG-QoL 15r scores at baseline and at Weeks 12, 24, and 60, are presented in Supplemental Figure 2.
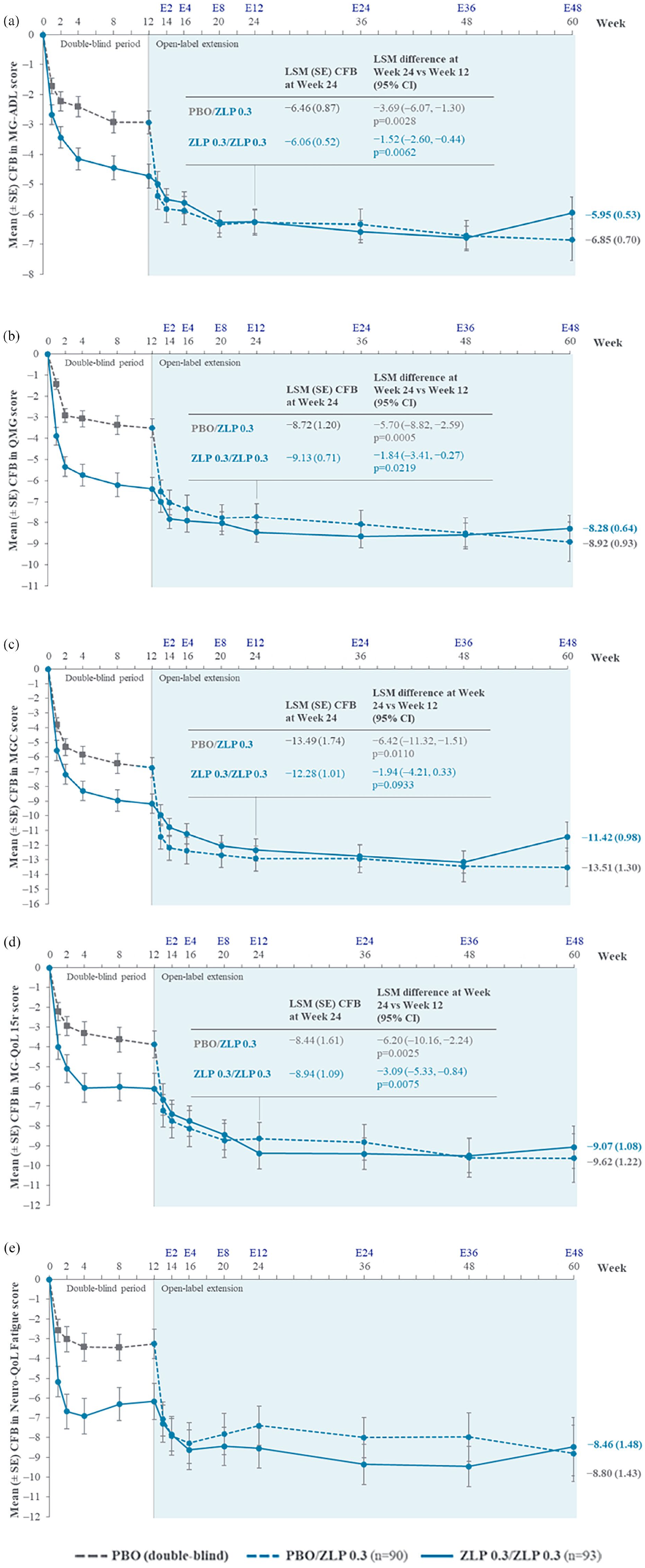
Change from DB baseline in (a) MG-ADL, (b) QMG, (c) MGC, (d) MG-QoL 15r, and (e) Neuro-QoL fatigue* scores up to Week 60.
In the PBO/ZLP 0.3 group, MG-ADL and QMG responder rates at Week 12 increased rapidly at Week 13 (1 week after switching to zilucoplan at Week 12), increased further through to Week 24, and were sustained through to Week 60. In the ZLP 0.3/ZLP 0.3 group, MG-ADL and QMG responder rates also increased from Week 12 to Week 24 and were sustained through to Week 60 [Figure 3(a) and (b)]. In the PBO/ZLP 0.3 and ZLP 0.3/ZLP 0.3 groups, MSE responder rates at Week 12 (8% and 19%, respectively) increased through Week 24 (33% and 31%, respectively), and were sustained through Week 60 [39% and 35%, respectively; Figure 3(c)]. Data for other exploratory efficacy endpoints (WPAI:SHP, MGFA-PIS, and EQ-5D-5L) are reported in Supplemental Material (Supplemental Table 2, Supplemental Figure 3, and Supplemental Figure 4, respectively).

(a) MG-ADL (⩾3-point reduction from baseline), (b) QMG (⩾5-point reduction from baseline), and (c) MSE (MG-ADL score of 0 or 1) responder rates without rescue therapy*.
A total of 63 patients from US sites completed the first SIAQ assessment during the OLE, of whom 52 patients also completed a second assessment 2 weeks later. Overall, SIAQ scores indicated that patients had a positive experience with self-injection and were highly satisfied at both time points (Supplemental Figure 5).
After adjusting for exposure, the rate of rescue therapy use during the DB period was 31.19 events per 100 patient-years for patients receiving ZLP 0.3 (n = 101) and 78.16 for patients receiving placebo (n = 103). Patients who switched from placebo to zilucoplan experienced a substantial decrease in rescue therapy use during the OLE (33.11 events per 100 patient-years) compared with the DB period (78.16 events per 100 patient-years). Thus, switching to zilucoplan in the OLE reduced the rate of rescue therapy by almost 60%, compared with the DB period. During the OLE, rate of rescue therapy use overall was 26.45 events per 100 patient-years. Approximately 15% of patients (n = 14 in both PBO/ZLP 0.3 and ZLP 0.3/ZLP 0.3 groups) had received rescue therapy during the OLE by the time of clinical data cut.
Among patients in the ZLP 0.3/ZLP 0.3 group who received corticosteroids (CS) at baseline and completed Week 60 at data cut off (n = 44), 41% (n = 18) patients discontinued or reduced CS dose relative to the DB baseline (mean DB baseline dose = 21 mg), with a mean CS dose reduction of 14 mg. In the PBO/ZLP 0.3 group, 41% (n = 12) of patients who received CS at baseline and completed Week 60 (n = 29) discontinued or reduced CS dose relative to the DB baseline (mean DB baseline dose = 27 mg), with a mean CS dose reduction of 16 mg. In the overall population, only 7 (12%) and 4 (7%) patients in the ZLP 0.3/ZLP 0.3 and PBO/ZLP 0.3 groups, respectively, increased CS dose up to Week 60 relative to DB baseline values; the mean CS dose increase was approximately 12 mg in both groups (mean DB baseline dose = 18 mg and 3 mg, respectively). Overall, mean MG-ADL and QMG score reductions at Week 60 were similar in patients who discontinued, decreased, or increased CS in both treatment groups.
PD and immunogenicity analyses
Complete complement inhibition was observed after 1 week (first assessment) of zilucoplan 0.3 mg/kg in the PBO/ZLP 0.3 group and was sustained through Week 60 (Supplemental Figure 6), following a similar trend to the complete complement inhibition at Week 1 observed in RAISE for the zilucoplan 0.3 mg/kg group. 10 Small numbers of low, positive ADA titers were reported in both treatment groups overall (n = 5, PBO/ZLP 0.3; n = 4, ZLP 0.3/ZLP 0.3). There was no evidence of an association between positive ADA status and reduced efficacy or incidence of adverse events.
Discussion
Zilucoplan is a small, 15-amino-acid macrocyclic peptide, which allows for simple daily self-administration via subcutaneous injection. This interim analysis of RAISE-XT showed that long-term treatment with zilucoplan had a favorable safety profile and was well tolerated in patients with AChR+ gMG. No new safety concerns were identified since the phase III study of zilucoplan, and the pattern of overall and serious TEAEs was similar to that observed in RAISE. 10 Notably, longer exposure to zilucoplan did not lead to higher rates of TEAEs overall.
MG worsening occurs as a result of disease fluctuations, but can also be triggered by factors such as infection, stress, or medications and supplements.22–24 During the OLE, only approximately a quarter of all patients had a TEAE of worsening of MG. Less than half (42%) of these patients required rescue therapy as deemed necessary by the investigator, suggesting that investigators were comfortable with a less aggressive treatment approach to manage disease fluctuations in the majority of patients. In addition, the use of rescue therapy decreases as time on zilucoplan increases, thus showing a positive effect of zilucoplan on the prevention of unpredictable gMG disease fluctuations. Furthermore, patients who received placebo in the DB period experienced almost a 60% decrease in rescue therapy use after switching to zilucoplan. Unlike monoclonal antibody C5 inhibitors, zilucoplan can be used concomitantly with IVIg and PLEX as rescue therapy, without the need for supplemental dosing.10,25,26
COVID-19 was the second most common TEAE reported during RAISE-XT and, in accordance with guidance from the International MG/COVID-19 Working Group, 27 it was recommended that patients who tested positive for COVID-19 did not stop receiving zilucoplan during RAISE-XT. While infections including COVID-19 can often exacerbate symptoms in patients with MG who are often immunocompromised due to treatment, the risk of stopping immunotherapy is also high. 28 Indeed, there are limited data to suggest that complement inhibition, including with zilucoplan, may even improve clinical outcomes of patients with COVID-19.29,30 There were no deaths related to COVID-19 in this study.
Overall, RAISE-XT demonstrated consistent and sustained improvement of gMG symptoms with zilucoplan across all efficacy endpoints assessed. Importantly, this sustained efficacy allowed for tapering or discontinuation of concomitant CS. This ability for patients to reduce or discontinue concomitant CS with zilucoplan reduces their risk of exposure to the systemic side effects and long-term toxicities that are associated with CS use. 31 In addition, MG-ADL and QMG responder rates increased over time to Week 60, suggesting that some patients will need more time to respond to zilucoplan. This pattern is also observed in the OLE studies of eculizumab and ravulizumab in patients with gMG,32–35 but the reasons for why late response occurs in some patients are not yet known. 2 However, the RAISE-XT data demonstrate that long-term treatment with zilucoplan enables more patients to achieve a clinically meaningful outcome beyond the DB 12-week period.
Zilucoplan also improved fatigue, an important outcome for patients that can affect everyday living, as demonstrated by a rapid and sustained improvement in Neuro-QoL Short Form fatigue scores. In addition, the absolute changes in MG-ADL, QMG, MGC, and MG-QoL 15r scores from DB baseline to Week 60 were of high magnitude and are greater than those observed over a similar timeframe for ravulizumab, also in a broad mild-to-severe gMG population. 8
There are several benefits of subcutaneous self-injection, compared with intravenous administration, including a reduced need for traveling to hospitals or clinics, reduced interference with daily plans and activities and greater independence, and avoiding the difficulties and complications associated with venous access.36,37 Zilucoplan has the added benefit of being a daily medication, which can help to reduce the peaks and troughs in efficacy that may be associated with less regular infusions, and it can also be stored at room temperature for up to 3 months, which can facilitate storage at home and while traveling. 38 However, some barriers remain, such as dexterity problems or injection anxiety.36,37 While the acceptability of daily self-injected subcutaneous zilucoplan was already suggested by the low discontinuation rate observed in RAISE and the high proportion of patients choosing to continue zilucoplan and enroll in the OLE, the consistent scores above 8 in the majority of SIAQ domains indicate a high patient satisfaction and a positive experience with self-injection among patients in the United States.
RAISE-XT has enabled the investigation of the safety and efficacy of zilucoplan beyond the 12-week DB period in a broad population of patients with AChR+ gMG. All patients who completed RAISE opted to enroll into RAISE-XT, and at the time of the data-cut, the large majority of patients were still enrolled with no discontinuations expressed as being due to lack of efficacy by the investigators. There are, however, some limitations to this study. RAISE-XT was designed to include patients from two randomized DB studies, each with their own inclusion criteria and prespecified efficacy and safety assessments, which resulted in some minor discrepancies when rolling over into the OLE study. For example, patients enrolled from the phase II study had lower average MG-ADL baseline scores (due to a lower MG-ADL score inclusion criterion) than those from the phase III study. However, results at Week 24 were adjusted by baseline MG-ADL score, so any impact on the overall outcome would be limited, and a post hoc analysis showed that results are consistent, whether patients from the phase II study were included or not (data not shown). In addition, the phase II study did not assess certain exploratory efficacy endpoints (e.g. Neuro-QoL) or ADAs, and therefore, data for patients enrolled from the phase II study are not available for the DB phase (up to Week 12) for these endpoints. Finally, RAISE-XT is ongoing and, at the time of the data-cut, some patients had not yet reached Week 60 and were, therefore, not included in the efficacy analysis at this time point.
Conclusion
Zilucoplan demonstrated a favorable long-term safety profile and was well tolerated in RAISE-XT, with no new safety concerns identified, and consistent efficacy in multiple endpoints that was sustained for up to 60 weeks. These data are in line with the rapid and clinically meaningful improvements observed after 12 weeks of zilucoplan treatment in RAISE.10,11 RAISE-XT is ongoing, and additional long-term data will be available in future analyses.
Supplemental Material
sj-docx-1-tan-10.1177_17562864241243186 – Supplemental material for Long-term safety and efficacy of zilucoplan in patients with generalized myasthenia gravis: interim analysis of the RAISE-XT open-label extension study
Supplemental material, sj-docx-1-tan-10.1177_17562864241243186 for Long-term safety and efficacy of zilucoplan in patients with generalized myasthenia gravis: interim analysis of the RAISE-XT open-label extension study by James F. Howard, Saskia Bresch, Constantine Farmakidis, Miriam Freimer, Angela Genge, Channa Hewamadduma, John Hinton, Yessar Hussain, Raul Juntas-Morales, Henry J. Kaminski, Angelina Maniaol, Renato Mantegazza, Masayuki Masuda, Richard J. Nowak, Kumaraswamy Sivakumar, Marek Śmiłowski, Kimiaki Utsugisawa, Tuan Vu, Michael D. Weiss, Małgorzata Zajda, Jos Bloemers, Babak Boroojerdi, Melissa Brock, Guillemette de la Borderie, Petra W. Duda, Mark Vanderkelen and M. Isabel Leite in Therapeutic Advances in Neurological Disorders
Footnotes
Acknowledgements
The authors thank the patients and their caregivers, in addition to the investigators and their teams, who contributed to this study. A full list of RAISE-XT study investigators is in Supplemental Table 3. The authors thank Holger Moeltgen and Iska Krautz of UCB Pharma for project management of the RAISE-XT study. Medical writing support was provided by Rachel Price, PhD CMPP of Ogilvy Health, London, UK, and funded by UCB Pharma, in accordance with Good Publications Practice (GPP3) guidelines (![]() ). The authors thank Veronica Porkess, PhD, CMPP, of UCB Pharma for publication and editorial support, and Natasa Savic, MD, PhD, and Hiroshi Todaka, PhD of UCB Pharma for critical review. The work conducted in Sheffield, UK, was supported by the UK National Institute for Health and Care Research Sheffield Biomedical Research Centre (IS-BRC-1215-20017).
). The authors thank Veronica Porkess, PhD, CMPP, of UCB Pharma for publication and editorial support, and Natasa Savic, MD, PhD, and Hiroshi Todaka, PhD of UCB Pharma for critical review. The work conducted in Sheffield, UK, was supported by the UK National Institute for Health and Care Research Sheffield Biomedical Research Centre (IS-BRC-1215-20017).
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.