
Abstract
Stroke is ranked as the second leading cause of death worldwide and a major cause of long-term disability. A potential therapeutic target that could offer favorable outcomes in stroke is the mammalian target of rapamycin (mTOR) pathway. mTOR is a serine/threonine kinase that composes two protein complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2), and is regulated by other proteins such as the tuberous sclerosis complex. Through a significant number of signaling pathways, the mTOR pathway can modulate the processes of post-ischemic inflammation and autophagy, both of which play an integral part in the pathophysiological cascade of stroke. Promoting or inhibiting such processes under ischemic conditions can lead to apoptosis or instead sustained viability of neurons. The purpose of this review is to examine the pathophysiological role of mTOR in acute ischemic stroke, while highlighting promising neuroprotective agents such as hamartin for therapeutic modulation of this pathway. The therapeutic potential of mTOR is also discussed, with emphasis on implicated molecules and pathway steps that warrant further elucidation in order for their neuroprotective properties to be efficiently tested in future clinical trials.
Introduction
Ischemic stroke is a severely debilitating and life-threatening neurological disorder characterized by disturbed cerebral blood supply that results into cerebral hypoperfusion and – ultimately – neuronal cell death. Worldwide it is ranked as the second leading cause of death and a major cause of long-term disability.1,2 Stroke is categorized in two main types, ischemic and hemorrhagic. 3 Because of the wide variety of acute ischemic stroke (AIS) causes, several classification schemes have been proposed based on the underlying etiology, including the TOAST (Trial of Org 10172 in Acute Stroke Treatment) classification system, that distinguishes between five different AIS types: cardioembolic, thromboembolic, lacunar, cryptogenic, and AIS due to other causes. 4
The mainstay treatment for ischemic stroke consists today of intravenous thrombolysis and endovascular thrombectomy, both aiming at recanalization of the occluded vessel and reperfusion of the ischemic cerebral tissue.5,6 The secondary purpose of acute stroke treatments is to mobilize any agents which can alleviate the damage which has already been inflicted upon the nervous tissue. Despite tremendous advances in the field of stroke therapies, time to reperfusion remains the main limiting factor. With this in mind, neuroprotective agents that will attenuate neuronal damage until vessel recanalization may be achieved while also preventing a potential reperfusion injury have recently come into the focus of stroke research. An attractive target for such agents seems to be the mammalian target of rapamycin (mTOR) pathway, which is involved in both autophagy-related apoptosis and inflammation, processes of equally great importance in stroke pathophysiology.
Stroke pathophysiology and the role of autophagy and inflammation
To gain insight into the pathophysiological importance of the mTOR pathway in stroke, the complex pathophysiological processes implicated in cerebral ischemia must first be elucidated.
The chain of events leading to neuronal cell death in ischemic stroke starts with a decrease in cerebral blood flow within a certain area of the brain. This propagates a cascade of cellular and molecular events, known as the ischemic cascade. 7 During the initial stages of ischemia, glucose- and oxygen-deprived neurons are forced into anaerobic metabolism. This, being an inherently less efficient energy production mode, results into a significant decrease in adenosine triphosphate (ATP) production, while at the same time, triggers release of lactic acid as a byproduct. 6 Subsequently, ATP-dependent ion transport pumps fail, causing the cell membranes to become depolarized and leading to a large influx of Ca+2. Intracellular Ca+2 levels are further increased through the release of glutamate, an excitatory neurotransmitter that binds to and opens Ca+2-permeable N-methyl-D-aspartate receptors. 8 The result of these cascades is activation of lytic enzymes and formation of free radicals. 9 In this context, of great importance is the role of calpain, a calcium-activated cytosolic protease which cleaves a number of different cytoplasmic and nuclear substrates. 10 Cells affected by ischemia eventually lose their structural integrity with their membranes becoming permeable to all sorts of ions and toxic chemicals and their organelles rendered inoperative. The end result of ischemia is activation of apoptosis and cellular death. Of note, autophagy and inflammation are mechanisms with a prominent role throughout this process. Triggered initially as processes for clearance of necrotic cells and toxic debris, they soon become somewhat of a ‘liability’ propagating brain injury.
Autophagy is a lysosome-mediated process that aims to remove, when activated, misfolded or aggregated cytosolic contents such as those encountered in stroke-affected cells.11,12 Three types of autophagy are described in the literature, microautophagy, chaperone-mediated autophagy and macroautophagy. 13 Macroautophagy utilizes double membrane vacuoles called phagophores to transport degraded cytoplasmic material to lysosomes in a five-step process. 13 First, phagophores engulf their target molecules in a process known as enucleation. Subsequently, this turns them into autophagosomes, organelles that will ultimately merge with lysosomes creating autolysosomes. 14 Autophagosomes have been identified in the hippocampus and also in the penumbra of stroke test animals and their importance in stroke pathophysiology has been well-documented. 15 In particular, the formation of autophagosomes is induced by upregulation of microtubule-associated protein 1 light chain (LC3)-II (Figure 1). Two different forms of LC3 exist (Figure 1). The cytosolic type of LC3 (LC3-I) is conjugated to phosphatidylethanolamine and forms LC3-phosphatidylethanolamine conjugate (LC3-II), which is responsible for the development of autophagosomal membrane 16 (Figure 1). In turn, within autolysosomes, enucleated macromolecules are eliminated through enzymatic cleavage. In the next phases which follow, elongation and expansion of the phagophore take place. Finally, through the transport of proteins to the lysosome the maturation of phagosome is achieved.17,18 This whole process is regulated through sophisticated signaling pathways, namely those of mTOR and adenosine-monophosphate activated protein kinase (AMPK)19,20 (Figure 1). mTOR inhibits autophagy by phosphorylating Unc-51-like kinase (ULK) 21 (Figure 1). AMPK on the other hand promotes autophagy by suppressing mTOR while also directly activating ULK to induce it. 22 It should be noted that ischemia propagates autophagy through AMPK activation23,24 (Figure 1).

Regulation of the autophagic process.
Inflammation also plays an important role in stroke, involving a multitude of molecular pathways and different types of inflammatory cells that are mobilized in the process. 25 Following an ischemic event in the cerebral parenchyma, there is increased expression of matrix metalloproteases (MMPs), such as MMP-2 and MMP-9, which lead to blood–brain barrier breakdown. 26 In addition, necrotic cells release molecules termed damage-associated molecular patterns (DAMPs), which mobilize the innate immune responses. Among the DAMPs which are released in the ischemic area, the high-mobility group protein 1 (HMGB1) is of special interest. 27 HMGB1 recruits microglial cells via membrane receptors called toll-like receptors (TLRs). 27 Microglial activation is elicited mainly by TLR2 and TLR4 but also by Advanced Glycosylation End Products (RAGE). 28 The end-result is release of cytokines, such as tumor necrosis factor (TNF), interleukin (IL)-1β, IL-6, and IL-8.27,29 TLRs also mediate the discharge of reactive oxygen species (ROS) through Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) by interacting with nuclear factor-κB (NF-κB). 30 Besides microglia, astrocytes also play a crucial role in ischemia, often worsening brain injury, again through cytokines.31,32 In turn, cytokine release in the ischemic core leads to leukocytic infiltration, with neutrophils being the main cells attracted in the affected region. This chain of events is mediated by expression of endothelial adhesion molecules by neutrophils which are attracted by a peptide called endothelin-1 that controls endothelial homeostasis. 33 Neutrophilic infiltration correlates with stroke severity and the extent of infarct.34,35 In this sense, molecular pathways that participate in the regulation of stroke-related inflammatory processes, including that of mTOR may play a key role in stroke outcomes.
The mTOR pathway
mTOR is a serine/threonine kinase, member of the phosphatidylinositol 3-kinase-related kinase family of protein kinases and a key regulator of cell growth. As already mentioned, mTOR modulates both autophagy and inflammation, playing an integral role in stroke pathophysiological processes.36,37 It is the core component of two separate protein complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). 36 mTORC1 consists of mTOR, regulatory-associated protein of TOR (RAPTOR), proline-rich AKT1 substrate of 40 kDa and mammalian lethal with SEC13 protein 8 (mLST8).38,39 On the other hand, mTORC2 consists of mTOR, mLST8, RICTOR (rapamycin-insensitive companion of mTOR), mSIN1 (mammalian stress-activated protein kinase-interacting protein) and protein observed with rictor-1.39–41 Dishevelled, EGL-10, and pleckstrin (DEP) domain-containing mTOR-interacting protein has been recently discovered as a negative regulator of both mTORC1 and mTORC2. 42
mTORC1 and mTORC2 are located in different subcellular compartments and serve several distinct functions. mTORC1 is found at the lysosomal surface where it controls protein synthesis and cellular growth in response to nutrients. 38 Its activity is regulated by various molecules (such as insulin, rapamycin, growth factors, neurotransmitters, and certain amino acids) as well as mechanical stimuli and oxidative stress.43–45 mTORC2, on the other hand, has been shown to regulate the actin cellular cytoskeleton and also to control cell metabolism, proliferation, and survival via phosphorylation of the protein kinases A, G, and C (AGC) family of kinases (namely, the serine/threonine protein kinase Akt/PKB). 46 In the human brain, mTOR is expressed in both glial cells and neurons. Specifically, mTORC1 plays a critical role in the development and differentiation of glia, especially microglia from oligodendrocytes. It is also responsible for lipid production in oligodendrocytes and Schwann cells and therefore essential for myelin synthesis.47–49 Moreover, mTOR plays a significant role in physiological functions of brain cells like memory, learning, plasticity, and sociability50–53 (Table 1).
Physiological functions of mTORC.

mTORC, mammalian target of rapamycin complex.
Dysregulation of mTOR function can lead to the development of neurodegenerative diseases, which are characterized by the aggregation of proteins. 54 Such is Parkinson’s disease, which is characterized by reduced dopamine in the striatum caused by abnormal aggregates of a-synuclein, which are named Lewy Bodies. It has been shown that the accumulation of misfolded a-synuclein is provoked by distorted autophagy through an mTOR-dependent pathway. 55 Huntington’s disease is an another neurodegenerative autosomal dominant disorder characterized by abnormal accumulation of a protein named huntingtin. It has been demonstrated that huntingtin induces the pathogenesis of this disease by promoting mTORC1 action. 56 In addition, mTORC1 is implicated in the pathogenesis of Alzheimer’s disease (AD), the most common cause of dementia, which is characterized by abnormal aggregation of amyloid-beta peptides and hyperphosphorylated tau that create amyloid-plaques and neurofibrillary tangles. 57 In animal models of AD and Huntington disease, the application of an mTORC1 inhibitor (rapamycin) reduced the toxic aggregates of proteins by promoting the processes of autophagy.56,57 Finally, another genetic disorder with dysregulation of mTORC1 activity is tuberous sclerosis complex (TSC). This is an autosomal dominant disorder and the majority of patients (85%) with this disease harbor mutations in TSC1 or TSC2 genes.58,59 As a multisystemic disorder TSC is characterized by the creation of hamartomas in various organs such as central nervous system (CNS), eyes, kidneys, skin, respiratory and circulatory systems.59,60 The most commonly affected system in TSC is CNS. In brain magnetic resonance imaging, anatomical deformities can be identified such as cerebral cortical tubers, subependymal nodules, and subependymal giant cell astrocytomas. These anomalies contribute to neurological symptoms such as epilepsy, autism and mental retardation, which are highly prevalent among TSC patients.
Besides neurodegenerative diseases, mTOR holds a crucial role in neuronal ischemia. In stroke, there is recruitment of various inflammatory cells (e.g. microglia, astrocytes) in the brain, which can have anti-inflammatory or proinflammatory actions. 61 Besides inflammation, autophagy is also implicated in stroke. 62 The common pathophysiological denominator between these two processes is the mTOR kinase.
mTOR and stroke
In this section, the role of the mTOR kinase in stroke through distinct processes of inflammation and autophagy will be discussed.
mTOR and inflammation
As already noted, mTOR-dependent regulation of the inflammatory process in stroke is particularly complex. In order for us to comprehend it, we need to elucidate the role of all implicated inflammatory cells. In general, mTOR, via its excitatory or inhibitory effects, is the main determinant of the outcome of post-stroke inflammation.61,63
Microglia
Microglial cells, activated through DAMPs, play a central role in the early stages of stroke-related inflammation acquiring either a proinflammatory (M1) or anti-inflammatory (M2) phenotype. 61 M1-type microglia cells promote inflammation via IL-1 and TNF-a, whereas M2-type cells block it through IL-10 and TGf-β (with the latter exerting their role via stabilization of the blood–brain barrier).61,64,65 mTORC1 has been shown to influence differentiation of microglia into an M1 or M2 phenotype. 66 Applying mTORC1 inhibitors in the acute phase of stroke, such as sirolimus or everolimus, diminished proinflammatory cytokines and promoted an anti-inflammatory M2 phenotype of microglia in experimental animal stroke models. 66 In addition, significant reduction of the infarct area and improvement in the behavioral tests of the experimental animals were noted. 66 Recent studies have also shown that administration of sestrin-2, a stress sensor protein, may elicit similar responses in experimental stroke models. In particular, administration of sestrin-2 induced microglial shift to an M2 phenotype and attenuated the ischemic brain injury by downregulating the mTOR pathway. 67 These findings align with experimental evidence that shows that the mTOR/STAT3 pathway is responsible for the regulation of proinflammatory cytokine production and induction of autophagy in microglia. 68 In accordance with the previous data, administration of 6-Gingerol, a natural compound isolated from ginger rhizomes, to rhodent stroke models has been shown to ameliorate microglia-induced neuroinflammation and cerebral injury by downregulating the Akt/mTOR/STAT pathway. 69 Analogous results were noted in the mTOR/STAT3 pathway by knockdown of the protein plexin-A2, which is responsible for regulating neuronal axonal guidance. Plexin-A2 knockdown facilitated M2 microglial polarization and increased functional recovery after ischemic stroke in experimental animal models. 70
Astrocytes and mTORC
Astrocytes are key modulators of immune responses in stroke. They may promote inflammation and tissue damage or contrarily, suppress inflammatory reaction acquiring a neuroprotective role. 61 The latter is achieved through reduction of glutamate dependent excitotoxicity as well as post-ischemic neurotoxic ROS.71–75 mTOR seems to play a central role in all these events. Previous research in rat stroke models has shown that increased activation of astrocytic mTOR via the PI3K/Akt pathway increases production of vascular endothelial growth factor (VEGF) and brain-derived neurotrophic factor, thereby promoting angiogenesis and neuroprotection in acute cerebral ischemia. 76 On the other hand, studies in primary astrocytic cultures under oxygen–glucose deprivation indicate that decreased activity of mTORC1 promotes autophagy and reduces proinflammatory cytokines through decreased production of TNF-a and downregulation of the TSC pathway.77,78
Oligodendrocytes and mTORC
Oligodendrocytes are neuroglia cells that produce myelin in neuronal axons. They originate from oligodendrocyte precursor cells (OPCs) and express certain antigens such as ganglioside GD3 and platelet-derived growth factor alpha receptor subunit. 79 The procedure of myelination is very complex and mTOR has been reported to play a crucial role in it. This notion is supported by various in vivo experiments which have unraveled the function of mTORC1 and mTORC2 in myelin generation. For example, reduced myelination was observed in raptor-knockout mice, when mTORC1 action was diminished.80–84 Under ischemic conditions in the acute phase of stroke, there is evidence of OPCs mobilization in the penumbra and reduction of their number in the ischemic core. This phenomenon is closely related to the PI3K/Akt/mTOR pathway. 85 Previous research has also demonstrated that inflammatory cascades induced by microglial activation may impair oligodendrocytes/OPCs, while the production of VEGF-C promotes oligodendrocytes/OPCs mobilization. 86 Oligodendrocytes interact also with lymphocytes and especially T- regulatory cells (T-regs), which produce IL-6 and induce OPC differentiation after the early stage of ischemic stroke. 87 After the ischemic event, the interaction between oligodendrocytes and endothelial cells has a pivotal role for the process of myelination in the ischemic area. 88 In a mouse model of extended cerebral hypoperfusion, Endothelial Progenitor Cell Secretome promoted neovascularization, increased the production of myelin and the number of mature oligodendrocytes in the damaged area. This was evident by the increase of p-AKT and p-mTOR which regulated the differentiation of OPCs in the recovery phase of ischemic stroke. Therefore, this effect of oligodendrocyte augmentation was potentiated by the PI3K/Akt/mTOR pathway. 89
Endothelial cells and mTORC
The barrier between blood vessels and brain parenchyma consists of astrocytic endfeet processes, endothelial cells, pericytes, and neurons. 90 Endothelial cells are located at the internal surface of the brain capillary and are a key regulator of inflammatory processes, as they mediate the penetration of inflammatory cells from the periphery to the CNS. In ischemic stroke, there is reduced blood flow in endothelial cells and altered substance exchange in the blood–brain barrier leading those cells to a proinflammatory phenotype. 91 In recent literature, there is evidence that mTOR-dependent autophagy can secure endothelial cell function post-stroke. 92 In an AD mouse model, application of rapamycin, an mTOR inhibitor, restored cerebral blood flow and reduced amyloid β-protein deposition in cerebral blood vessels. 92 This mechanism was associated with endothelium-dependent vasodilation via nitric oxide production and eNOS (endothelial nitric oxide synthase) phosphorylation. 92 In another study, in stroke animal models, rapamycin was shown to enhance collateral perfusion in normotensive Wistar rats and spontaneously hypertensive rats, which were subjected to cerebral ischemia by middle cerebral artery occlusion. 93 In these animals, rapamycin improved collateral perfusion by enhancing eNOS activation in isolated and pressurized leptomeningeal anastomoses. In addition, rapamycin enhanced cerebral blood flow during reperfusion (i.e. immediately after stroke), resulting in significant reduction of the infarct volume and improved stroke outcome. In view of these findings, rapamycin is thought to comprise an attractive therapeutic agent in AIS, both improving the collateral flow and reperfusion outcomes (i.e. following thrombolysis or thrombectomy). 93 In another study, induction of autophagy in brain endovascular endothelial cells through an mTOR-dependent pathway promoted stabilization of blood–brain barrier integrity after oxygen glucose deprivation/reperfusion induction. 94
mTOR and autophagy
As already mentioned, mTORC1 is located at the cytoplasmic surface of lysosomes, the exact same organelles responsible for autophagic degradation. When activated, mTORC1 inhibits autophagy through two main mechanisms. First and foremost, it directly and rapidly blocks autophagosome formation at its very early stages through ULK phosphorylation95,96 (Figure 1). In addition, and this time via phosphorylation of transcription-factor-EB protein family members, it downregulates lysosomal proteins, ultimately restricting lysosomal biogenesis and function. 97 Of note, mTORC1 is blocked by AMPK, the second key player in autophagy modulation. AMPK promotes autophagy through phosphorylation of RAPTOR (and thus inhibition of mTORC1) but also through direct activation of ULK23,24 (Figure 1).
Moreover, mTOR restricts autophagy as part of mTORC2. Activation of mTORC2 leads to phosphorylation and activation of Akt and other members of the AGC kinase family 98 (Figure 2). This in turn results into downregulation of autophagy and lysosome-related proteins, mainly through regulation of FoxO transcription factors. 99
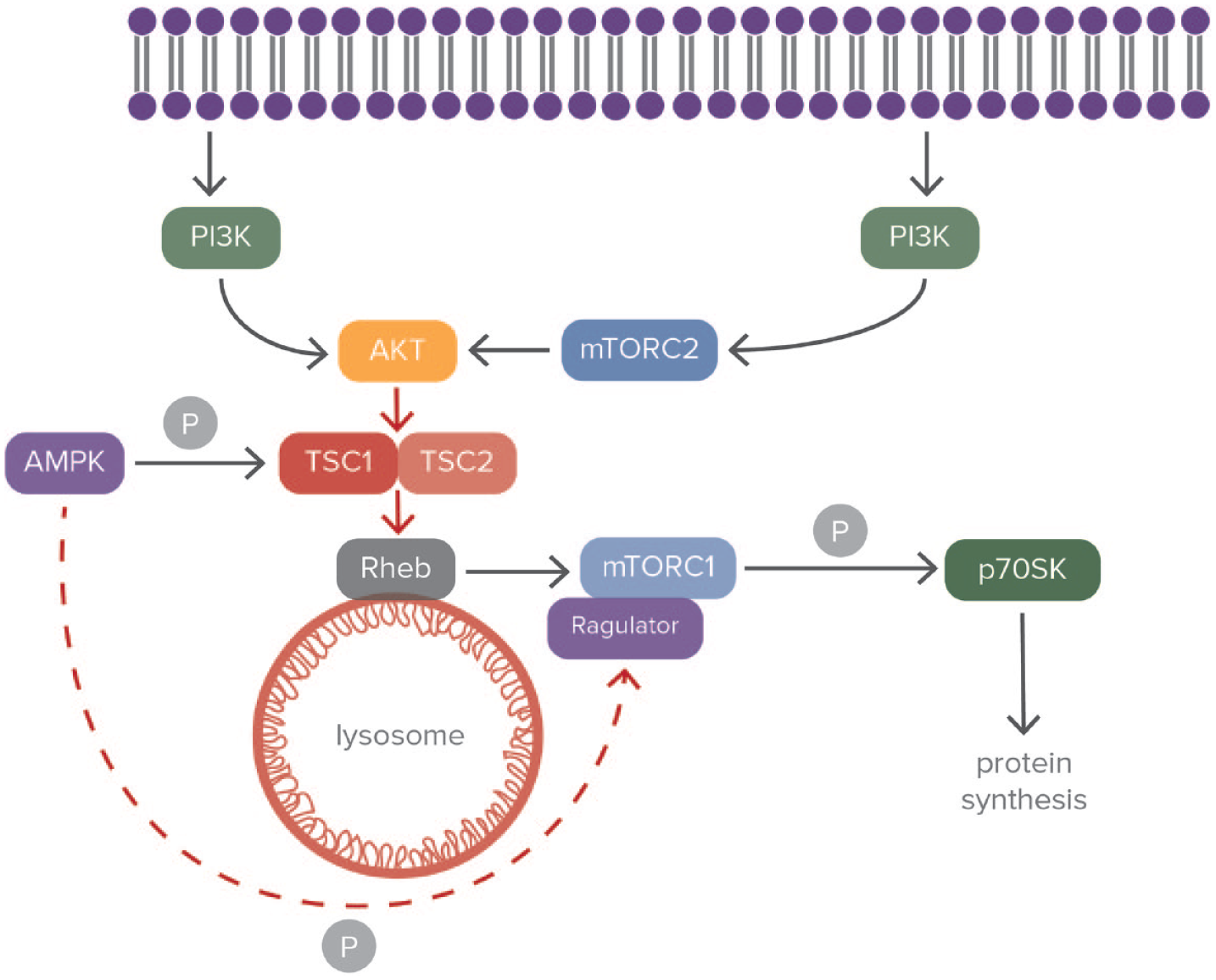
Modulation of tuberous sclerosis complex through the mTORC pathway.
Stroke has been shown to propagate autophagy through AMPK activation 17 (Figure 1). Nevertheless, whether this is beneficial or not for neurons is not clear. In PC12 cell line cells (derived from a transplantable rat pheochromocytoma), application of 3-methyladenine (3-MA), a well-known autophagy inhibitor, reduces cell apoptosis under ischemic conditions. 100 Along the same line of evidence, inhibition of autophagy either pharmacologically or by lentiviral vector delivering, accomplished reduction of infarct volume in stroke rat models. This was achieved by intraventricular 3-MA administration or by silencing autophagy promoters, Beclin1 and autophagy-related gene 7 (Atg7), via transferring short hairpin RNAs (shRNAs). 101 By contrast, other studies show that 3-MA and wortmannin (again an autophagy inhibitor) modulate rat ischemic neurons to transit from apoptosis to the necrotic phase. 102 Also, treatment of neuronal cells from neonatal rats with rapamycin under hypoxic conditions results into increased expression of the autophagy markers Beclin-1 and LC3-II along with reduced neuronal death. 102 Furthermore, hamartin, the protein end-product of TSC1, seems to increase in ischemic CA3 neurons (CA refers to Cornu Ammonis hippocampal areas), mediating neuronal survival via the pathway of autophagy. CA3 which is part of hippocampal structure is particularly resistant to ischemia. In order to identify the mechanisms responsible for this phenomenon, a proteomic analysis of this hippocampal region was performed in stroke rat models. It was found that hamartin was among the main proteins which are expressed following ischemia. This protein induced autophagy by blocking the mTORC1 pathway. 15
Targeting the mTOR pathway in stroke
The mTOR pathway has been introduced as potentially providing neuroprotection under ischemic conditions in stroke. 103 In neuronal and microglial cell cultures exposed to oxygen/glucose deprivation conditions, inhibition of mTOR signaling results in neuronal cell death.99,104 Moreover, experiments in fresh water invertebrates exposed to sodium hyposulfite revealed that metformin-induced mTOR upregulation acts protecting neurons from hypoxia. 105 Melatonin in stroke rodent models reduces infarct volume through mTOR and p70 S6 kinase activation (and thus Akt inhibition), while remote ischemic preconditioning in hippocampal cells increases phosphorylated mTOR and reduces apoptotic rate. 6 Finally, it has been demonstrated that Phosphatase and Tensin Homolog Deleted On Chromosome 10 suppression reduces cerebral infarct in middle cerebral artery occlusion through mTOR phosphorylation and activation. 106 However, not all published results support this neuroprotective role of mTOR. For example, in a recent publication involving transient forebrain ischemia in rats, mTOR inhibition with rapamycin resulted into apoptosis reduction and reduced ischemia-induced damage. 107
This discrepancy of mTOR inhibition and activation effects on stroke outcomes was highlighted in a recent systematic review and meta-analysis. 108 In this meta-analysis, 17 original studies were included, which evaluated the effect of rapamycin in animal stroke models. It was noted that even though rapamycin was neuroprotective in animal stroke models, the extent of its efficacy was influenced by the administered dose. This was attributed to the fact that rapamycin at high doses could be deleterious due to uncontrollable autophagy, whereas at low doses could be protective to neurons. 108 Another explanation for these discordant effects could be the suppression of mTORC2 due to higher dose or prolonged rapamycin treatment. 108 Moreover, alternative hypotheses have also been put forth, suggesting that the therapeutic manipulation of the mTOR pathway in stroke may be time-dependent (i.e. with mTOR inhibition generally considered beneficial in the acute phase of stroke, but its mobilization is potentially detrimental in the recovery phase of stroke when myelination and angiogenesis is required).66,89
TSC complex and stroke
Of certain interest is the close relation of the mTOR pathway with hamartin-tuberin complex under hypoxic conditions. 109 Hamartin connects with tuberin, which is the transcriptional product of TSC2 gene and creates a protein complex (the TSC1-TSC2 complex) 110 (Figure 2). Hamartin and tuberin are responsible for various cellular functions such as steroid hormone modulation, cell cycle regulation, and vesicular transport. Hamartin is essential for the stabilization of tuberin, since it inhibits its degradation. When the cell is subjected to ischemic conditions, there is activation of Rheb (Ras homolog enriched in brain) which is a guanosine triphosphate (GTP)-binding protein (Figure 2). Through this mechanism, there is blocking of mTORC1 by the TSC complex. This procedure leads to adjustment of p70S6 K (ribosomal protein S6 kinase), 4E-BP1 (eukaryotic translation initiation factor 4E-binding protein 1), and eEF2 K (eukaryotic elongation factor 2 kinase), which are all essential for cell maturation and protein production 111 (Figure 2). Moreover, the formation of the TSC complex is modulated by the cyclin-dependent kinase 1 (CDK1), while AKT modulates its dispersion by phosphorylating hamartin.
The hippocampus proper is a hippocampal structure that consists of the four segments CA1, CA2, CA3, CA4. It has been proved that CA3 cells are particularly resistant to ischemic insult whereas CA1 cells exhibit a relative sensitivity to such conditions.112–115 This heterogeneity attracted scientific attention and proteomic analysis was performed in this structure under hypoxic conditions. These investigations established that hamartin is upregulated in CA3 cells under hypoxic conditions while in CA1 cells its expression remained unaffected. Particularly in CA1 neurons, which remained unaltered by ischemia, there was increased expression of this protein only when these cells were exposed to ischemic preconditioning. 116
Neuroprotection can be accomplished by hamartin through the modulation of the autophagic process. 15 As noted above, autophagy is a homeostatic mechanism vital for cellular demands. The cell can utilize autophagy for the degradation of macromolecules in order to support its energy demands. 117 Global ischemia in CA1 neurons of hippocampus promotes production of membranous structures called autophagosomes due to diminished autophagy. Autophagosomes play a vital role in the process of macroautophagy. 118 However, modulation of autophagy through the application of rapamycin (an mTOR inhibitor) elicited neuroprotective effects on neurons. In a previous study, a TSC1 shRNA viral vector was utilized in order to suppress the expression of hamartin in vivo and in vitro. In this way, neurons became more sensitive to ischemic conditions and entered apoptosis. In this study, the researchers also showed that by increasing the expression of hamartin by a lentiviral vector in hippocampal neurons, they increased their resistance to oxygen glucose deprivation conditions. This was possible by inducing the autophagic process by blocking the mTORC1 pathway. Also in vivo, the rats that were stereotactically injected with the TSC1 shRNA viral vector exhibited increased locomotor activity post-ischemia.15,119
As already noted, hamartin may be a promising therapeutic agent in various diseases including stroke. Since its neuroprotective potency was proven in in vivo studies in hippocampal neurons, its potential effect in different neuronal cells that are responsible for major disabilities in humans such as cells of the primary motor cortex still remains to be established. Apart from neuronal cells, the effect of hamartin on inflammatory cells also remains to be ascertained. In 2020, another group of researchers demonstrated that increased expression of TSC2 in astrocytic cultures under oxygen/glucose deprivation conditions, resulted into increased autophagy by reducing the phosphorylation of mTOR. This was possible via the application of dexmedetomidine (a sedative agent), which reduced apoptosis in the astrocytic culture. 78 Therefore, the effect of hamartin in different cell types such as glial cells should be thoroughly investigated in the future.
Although such therapies are still not clinically available, ongoing research is expected to soon shed light onto the therapeutic potential of novel molecules including exosomes for protein modulation such as hamartin. 120 Exosomes are extracellular vesicles, which are secreted by all cells and contain microRNAs. 121 By producing engineered exosomes, these would be able to cross the blood–brain barrier, due to their small size, and mediate intracerebral production of hamartin.
Conclusion
The last two decades have witnessed an increase in the incidence of stroke by 70%. 122 It is estimated that stroke prevalence and incidence among people <70 years old have increased by 22% and 15%, respectively.123–125 Clearly, our main goal when treating stroke is to restore cerebral blood flow to the ischemic area, reducing the damage inflicted upon neurons due to oxygen deprivation. This is achieved by acute recanalization therapies, either by intravenous thrombolysis with recombinant tissue-type plasminogen activator or by endovascular thrombectomy. The latter is today the mainstay treatment for stroke based on the results of a number of clinical trials that have been published since 2015 (e.g. MR CLEAN, EXTEND-IA, SWIFT PRIME) proving its efficacy and safety.126–131 Nevertheless, recanalization therapies cannot guarantee a good functional outcome, with time-to-therapy being the main limiting factor. 131 The development of neuroprotective agents, which can be implemented as add-on treatments to delay neural death until vessel recanalization may be achieved, are hence of paramount importance. Such an attempt was the ESCAPE-NA1 trial (Efficacy and safety of nerinetide for the treatment of AIS), which was a multicenter, double-blind, randomized, placebo-controlled, phase III trial to test a neuroprotective agent (nerinetide) in the acute phase of stroke. 132 Even though the results of this trial were negative, the era of neuroprotective agents in stroke is still at its dawn.
A promising target for neuroprotective interventions is the mTOR pathway, the common underlying pathway regulating ischemia-related inflammation and autophagy. Promoting or inhibiting such processes under ischemic conditions can lead to apoptosis or instead sustained viability of neurons. Therefore, the utilization of agents that can regulate inflammatory and autophagy cascades in the brain aims to attenuate neuronal apoptosis and increase neuronal survival in stroke.
To conclude, future research is direly needed to assess the full neuroprotective and therapeutic potential of the mTORC pathway in ischemic stroke. The key notion of this approach lies in the mobilization of this pathway via neuroprotective agents such as hamartin that allow ‘calibrated’ interactions with the mTORC pathway, rather than simplified approaches that attempt initiation or suppression of this pathway as a whole. In the latter case, as complex cascades are induced at neuronal level, the completion or contrarily the premature termination of these processes is hard to ascertain. Thus, when modulating the mTORC pathway, in vivo cautious interpretation of experimental findings is warranted in order to avoid erroneous inferences. For example, the role of mTORC may be deemed crucial under the premise that the pathway initiation was completed, when in reality, it could be the premature termination of these processes, which led to neuronal apoptosis under ischemic conditions.