
Abstract
A multitude of environmental factors can result in breakdown of immune tolerance in susceptible hosts. Infectious pathogens are among the most important environmental triggers in the pathogenesis of autoimmunity. Certain autoimmune disorders have a strong association with specific infections. Several neurological autoimmune disorders are thought to occur through post-infectious mechanisms. In this review, we discuss the proposed mechanisms underlying pathogen-induced autoimmunity, and highlight the clinical presentation and treatment of several post-infectious autoimmune neurological disorders. We also highlight post-infectious neurological disorders in the setting of recent outbreaks.
Keywords
Introduction
Autoimmune disorders are caused by a breakdown of immune tolerance, which results in a misdirected response to self-antigens. Complex interactions between genetic, immunological, and environmental factors contribute to this loss of tolerance. 1 Infectious pathogens are an important environmental trigger in the pathogenesis of autoimmunity, as demonstrated by the strong association between certain autoimmune disorders with specific microorganisms (e.g., the association of Streptococcus and rheumatic fever).2,3 For most autoimmune disorders, the correlation with infection is more nuanced. A variety of pathogens have been implicated in the development of any single autoimmune disorder (Table 1), suggesting that infections may trigger autoimmunity through mechanisms beyond molecular mimicry. 4 The cumulative exposure to infections during childhood has also been proposed as an important factor in autoimmunity in adulthood. 2
Autoimmune neurological disorders and their associations with infections.

HIV, human immunodeficiency virus; SARS-CoV-2, severe acute respiratory syndrome coronavirus-2.
The role of infections in the pathogenesis of ‘classic’ neuroimmunological disorders such as multiple sclerosis and Guillain–Barre syndrome (GBS) has been studied extensively.5,6 Recent breakthroughs in biomarker discovery have led to the recognition of new antibody-mediated neurological disorders targeting neuronal and glial proteins. Infections are emerging as an important trigger for these novel autoimmune disorders, though the mechanisms behind loss of tolerance remain poorly understood. Here, we review the current landscape of post-infectious neurological autoimmunity, discuss proposed immunological mechanisms, highlight specific disorders strongly associated with pathogens, and review treatment considerations.
Proposed mechanisms of pathogen-induced autoimmunity and their association with neuroimmunological conditions
Molecular mimicry
Molecular mimicry (Figure 1) was initially described in 1964 as a mechanism through which pathogens evade the immune system, thus gaining evolutionary advantage. 7 Epitopes present on microorganisms may share marked similarity in peptide sequence or three-dimensional structure to host antigens, allowing a pathogen to establish immune tolerance. In susceptible hosts, homologous antigens on a pathogen may illicit an immune response; as a consequence, activated lymphocytes may aberrantly cross-react with self-antigens. Evidence suggests this misdirected response contributes to the pathogenesis of several systemic autoimmune disorders. 8
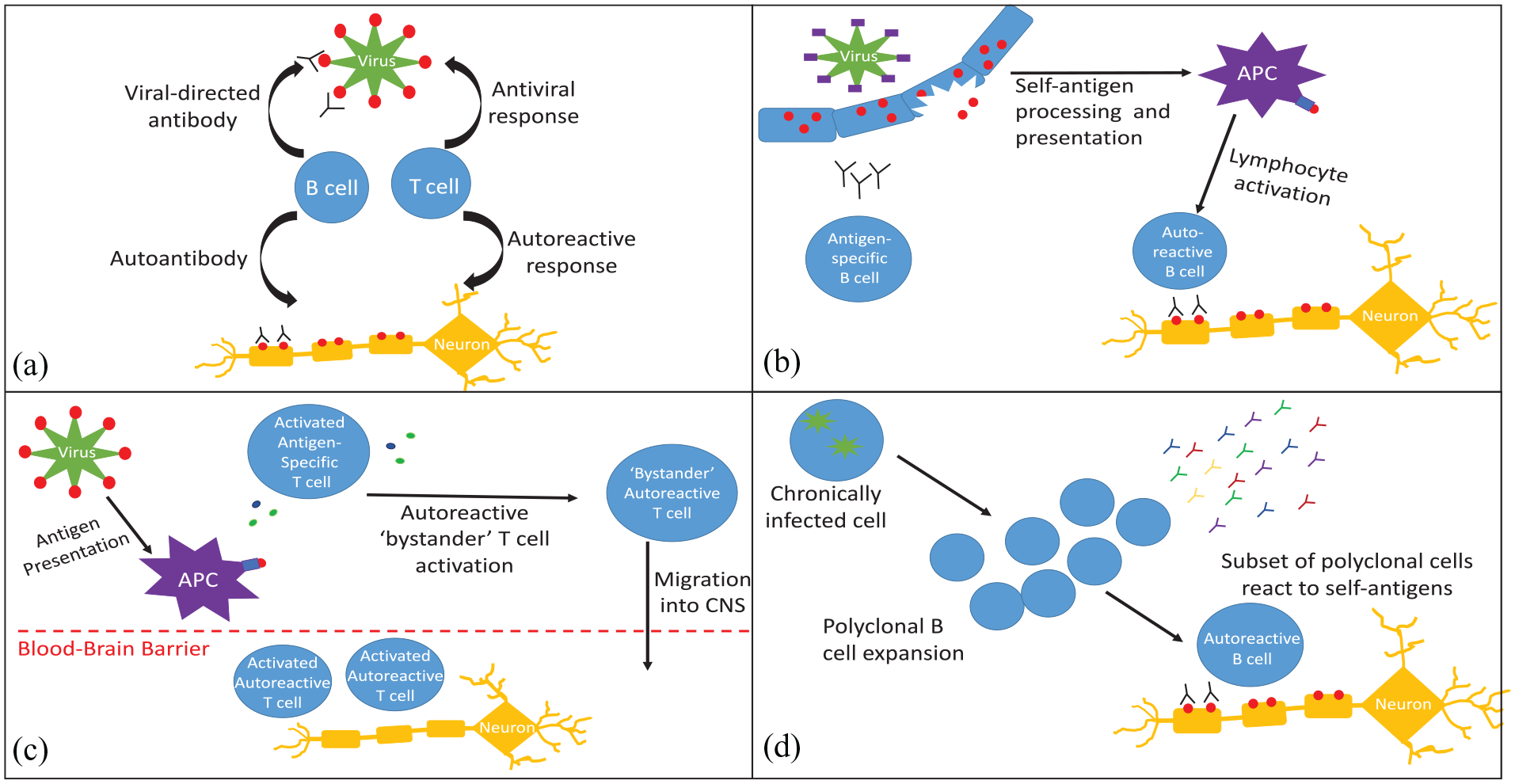
Mechanisms for loss of immune tolerance. (a) Molecular mimicry: an antigen present on a pathogen has a homologous structure to a self-antigen, resulting in loss of immune tolerance and inflammatory response to host antigens. (b) Epitope spreading: the initial response to acute infection is highly specific, but can broaden to other epitopes on the pathogen. This may eventually include self-antigens, resulting in autoreactive lymphocyte activation and autoimmunity. (c) Bystander activation: in response to an infectious pathogen, APCs, cytotoxic T Cells and helper T cells produce inflammatory mediators, which can activate autoreactive lymphocytes. Systemic inflammation can also cause blood-brain barrier disruption, granting autoreactive lymphocytes access to the CNS. (d) Persistent infection and polyclonal expansion: chronic infections, such as Epstein–Barr virus, may cause polyclonal B-cell expansion. A subset of these B cells may produce antibodies that react to self-antigens.
Molecular mimicry contributes to the breakdown of self-tolerance in certain neuroimmunological disorders, and may play a role in the pathogenesis of diseases such as multiple sclerosis and myasthenia gravis (MG). 9 Acute rheumatic fever serves as a classic example of mimicry-induced autoimmunity, and homology between epitopes on Group A Streptococcus (GAS) and brain gangliosides are believed to contribute to neurological sequelae of the disorder. 3 Mimicry is also important in axonal variants of Guillain-Barre syndrome (GBS), where similarities between a carbohydrate moiety on strains of Campylobacter jejuni result in autoantibodies targeting the human GM1 ganglioside (see section on GBS for details).6,10
Epitope spreading
The initial immune response to an antigen (host or non-host) is highly specific – typically limited to specific peptide sequences on the targeted protein. This response can gradually broaden to include different epitopes on the inciting antigen, or to new antigens altogether, a process known as epitope spreading (Figure 1). Mechanisms resulting in epitope spreading include post-translational modification of epitopes (such as substitution of arginine to citrulline), the release and presentation of ‘hidden’ antigens encountered during tissue injury, and somatic hypermutation.11,12 Epitope spreading may be an important mechanism in propagating neurological autoimmunity. The work of Miller and colleagues demonstrated evidence of myelin reactive T cells in the weeks following infection with Theiler’s murine encephalomyelitis virus (a mouse model of multiple sclerosis), demonstrating that viral infections may trigger autoimmunity via antigen release. 13
Bystander activation
While several mechanisms exist to maintain self-tolerance, evidence suggests that considerable numbers of autoreactive lymphocytes enter the periphery. 14 During an immune response to a highly virulent pathogen, lymphocytes may be activated via antigen-independent mechanisms. The concept of bystander activation maintains that such an inflammatory cascade may stimulate autoreactive immune cells, resulting in autoimmunity.14,15 Systemic inflammation can also result in dysfunction of the blood brain barrier, granting autoreactive cells access into the nervous system. 16 Bystander mechanisms (Figure 1) may play an important role in the development of CNS autoimmunity. A ‘dual signal’ hypothesis for the pathogenesis of MS proposes that autoreactive cells may be primed via mimicry or epitope spreading, but that a second inflammatory event results in lymphocyte activation and migration into the CNS. These complex interactions may explain why different pathogens have been implicated in the development of autoimmune disorders such as MS.17,18
Persistent infection and polyclonal B-cell activation
Certain infectious pathogens, such as herpesviruses or hepatitis C, are able to evade immune clearance and persist in the host indefinitely. A chronic inflammatory response resulting from persistent infection mediates a polyclonal proliferation of B and T cells (Figure 1). This process may result in the production of autoantibodies or immune complexes which contribute to the development of autoimmunity.4,19 Such mechanisms could explain the high prevalence of autoimmune disorders in patients with chronic hepatitis C infection. 20
Post-infectious autoimmune neurological disorders
Guillain–Barre syndrome
GBS is an immune-mediated polyradiculoneuropathy, and the most common cause of acute flaccid weakness worldwide.21,22 GBS typically presents with ascending limb weakness, sensory symptoms, and hyporeflexia, though other clinical features (such as cranial neuropathies, ataxia, or autonomic dysfunction) may be present. There are several recognized GBS “variants”, including acute inflammatory demyelinating polyradiculoneuropathy (AIDP), acute motor/sensory axonal neuropathy (AMAN or AMSAN) and Miller Fisher syndrome (with prominent ophthalmoplegia and ataxia), among other less common forms.21,22 GBS is a prototypical post-infectious neurological syndrome, with up to 70% of patients recognizing a preceding respiratory or gastrointestinal illness within 2 weeks of neurological symptom onset.21–23 Furthermore, serologic evidence of antecedent infection is found in a high proportion of GBS patients in case-control studies.24,25 Viruses frequently associated with the development of GBS include cytomegalovirus (CMV), Epstein–Barr virus, influenza, human immunodeficiency virus (HIV), and hepatitis A and E. GBS-associated bacterial infections include Campylobacter jejuni, Chlamydia pneumonia, Mycoplasma pneumonia, and Haemophilus influenzae.21,24–26 Recent outbreaks of arboviral infections and novel respiratory viruses have also resulted in an increased incidence of GBS in affected nations (see section on post-outbreak neurological complications for details).
Evidence suggests that GBS is predominantly an antibody-mediated disorder. Serum from patients with AIDP can cause demyelination of peripheral nerves and dorsal root ganglia in animal models. Furthermore, histologic investigation from patients with GBS has demonstrated the presence of antibodies and membrane attack complex on peripheral nerves, though to date the specific target of antibodies and their role in AIDP remain poorly understood.22,27,28 Antibodies targeting gangliosides on peripheral nerves (e.g., GM1, GD1a, GQ1b) have been identified in up to 80% of patients with axonal GBS and Miller Fisher syndrome.21,27,29 Animal studies have demonstrated pathogenic potential of these antibodies, with GM1/GD1a antibodies resulting in axonal damage, with GQ1b causing conduction block.27,30–32
Campylobacter jejuni is the organism most frequently associated with GBS, most commonly the AMAN and Miller Fisher variants. The relationship between this bacterium and GBS demonstrates a classic example of mimicry-induced autoimmunity.21,22,27 Certain strains of C. jejuni carry a sialtransferase gene, which is responsible for the expression of lipooligosaccharides (LOS) present on the bacterial cell wall.33–35 Portions of this LOS are homologous to human gangliosides, and substitutions within specific loci result in antigens that mimic GM1, GD1a, or GQ1b.33–35 In animal models, immunization with C. jejuni LOS mimicking GM1 or GD1b LOS produced an axonal neuropathy.6,33 Evidence of mimicry-induced axonal GBS in human was discovered inadvertently when injections containing gangliosides were administered for neuropathic pain, resulting in an acute axonal neuropathy with GM1 autoantibodies. 36
Mimicry-induced autoimmunity may play an important role in GBS cases associated with severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) infection. 37 The characteristic “spike” protein present on coronaviruses is capable of binding to receptors on the surface of respiratory cells, facilitating viral infection. While much attention is given to the role of the angiotensin-converting enzyme 2 (ACE-2) receptor in coronavirus transmission, the spike protein also interacts with cell-surface glycoproteins and gangliosides, including the GalNAc moiety on GM1. 37 Antibodies to GM1 and GD1b have been reported in cases of SARS-CoV-2 associated GBS, suggesting potential cross-reactivity between the spike protein and human gangliosides.37–39
Post-infectious autoimmune encephalitis
Herpes simplex virus encephalitis
Herpes Simplex Virus Encephalitis (HSE) remains a prominent cause of encephalitis worldwide, with high mortality despite early recognition and treatment with acyclovir. 40 Patients with HSE have a high rate of delayed neurological exacerbations within the first 8 weeks of treatment. In some cases, active viral replication is confirmed in the cerebrospinal fluid (CSF), consistent with recurrent HSE; however, a significant proportion of cases have negative viral testing and do not improve with antiviral agents.41,42 It was observed that some patients with post-HSE worsening improved with corticosteroids, suggesting that the underlying process may be immune-mediated. Further support for a post-infectious autoimmune encephalitis was discovered when neuronal autoantibodies were detected in the serum and CSF of post-HSE patients. 43
Among post-HSE patients, studies suggest up to 27% will subsequently develop autoimmune encephalitis. 44 Symptoms generally develop within 60 days of HSE, but cases occurring 2 years following initial infection have been reported. Two age-related clinical phenotypes have been identified in post-HSE autoimmune encephalitis. Children less than 4 years of age develop prominent choreoathetosis, often accompanied by impaired sensorium. Frequent seizures are also observed in this population, including infantile spasms, which may occur acutely or at later stages. Older children and adults experience prominent neurobehavioral manifestations (including psychosis) as an early manifestation, which may initially be attributed to residual deficits from HSE. 44
Neuronal antibodies are frequently detected in patients with post-HSE autoimmune encephalitis. Antibodies to the N-methyl-
Mechanisms underlying the development post-HSE AE remain debated. One study identified high rates of serum HSV positivity among patients with NMDA-R encephalitis that did not have clinical HSE, leading the authors to conclude that molecular mimicry may lead to the production of autoantibodies. 49 However, as HSV-positive patients may develop antibodies to antigens other than NMDA-R, an aberrant immune response may result from antigens being released during the inflammatory phase of HSE. In one longitudinal study, approximately half of post-HSE patients produced autoantibodies; however, only 56% percent of antibody positive patients went on to develop autoimmune encephalitis.44,46 Further study is needed to understand host and environmental risk factors that result in encephalitis in post-HSE patients with autoantibodies.
Japanese encephalitis
Japanese encephalitis (JE) virus is an important cause of encephalitis in Asia, with an estimated incidence of 69,000 cases per year. 50 JE is typically a monophasic disease, with headaches, fever, seizures, and altered sensorium, but rare cases of early relapse have been reported. 51 Similar to HSE, patients with post-JE worsening were noted to experience prominent dyskinesias and significant behavioral changes, leading investigators to explore an immune-mediated etiology in such cases. Further evidence emerged from a single-center analysis of three cases of post-JE relapse, and all had NMDA-R antibodies present in CSF. A follow-up study examined 65 JE patients prospectively, identifying relapse in five cases (7.9%). NMDA-R antibodies were present in three of these cases, while the other two were felt to have autoimmune encephalitis based upon negative JE virus testing and improvement with immunotherapy.52,53 Larger, multi-center studies of JE patients are needed to determine the incidence of post-JE autoimmune encephalitis, and to identify other autoantibodies that may be associated with the disorder.
Epstein–Barr virus and post-transplant autoimmune encephalitis
Rare cases of autoimmune encephalitis associated with solid organ and hematopoietic stem cell transplant have been reported in the literature. Reported cases have been associated with NMDA-R, AMPA, and myelin oligodendryocyte glycoprotein (MOG) antibodies. The mechanisms behind loss of tolerance despite immunosuppression associated with transplant are poorly understood, but an association with Epstein–Barr virus (EBV) has been proposed, with a few cases reporting positive EBV testing in the CSF corroborating an association.54–56
Sydenham’s chorea
Acute rheumatic fever (ARF) is a multifocal inflammatory condition that presents in the weeks following GAS infection. Sydenham’s chorea (SC) is a neurological manifestation of ARF, with an estimated prevalence ranging from 10% to 50%, which can present between 1 and 8 months following infection.57,58 SC is characterized by hyperkinetic, adventitious movements of the trunk and extremities, frequently occurring bilaterally. Other features, such as motor persistence, motor/vocal tics, and decreased tone have also been associated with the disorder.58,59 Behavioral alterations were previously underappreciated in the disorder, but recent studies have reported frequent emotional lability and obsessive compulsive behaviors. 60 SC was previously thought to be a self-limited disorder, but recent reports suggest a relatively high rate of recurrent chorea, many in the setting of acute streptococcal infection. 61
The first antineuronal antibodies isolated from individuals with SC were noted to react with caudate and subthalamic nucleus. Moreover, the presence of antineuronal antibodies correlated with the severity and duration of chorea. 62 Similar findings were reported with children diagnosed with SC at the National Institutes of Mental Health (NIMH) in whom anti-neuronal antibodies directed against human caudate tissue were demonstrated in 10 of the 11 patients. 63 In another study conducted by the NIMH, 24 patients were compared with age-matched controls and were found to have increased volume of caudate, putamen, and globus pallidus on brain magnetic resonance imaging, which may relate to acute inflammatory changes. 64
Treatment of suspected SC is multifaceted, and includes both symptomatic management and prophylaxis for the non-suppurative neurological and systemic sequelae of GAS. Recurrent ARF can lead to worsening of rheumatic heart disease; thus, prophylactic antibiotics are recommended in children with manifestations of ARF even in the absence carditis. Intramuscular Benzathine Penicillin G, administered every 4 weeks, is considered first-line for prophylaxis given long-standing efficacy and relatively narrow spectrum of activity. 65 Symptomatic treatment of chorea is necessary if they cause difficulty with activities of daily living, and generally involves the use of dopamine receptor blockers or anticonvulsants. In cases of severe or refractory disease, immunotherapies, including glucocorticoids, intravenous immunoglobulins, and plasmapheresis, have reportedly led to improvement. 3
Acute disseminated encephalomyelitis
Acute disseminated encephalomyelitis (ADEM) is an inflammatory CNS demyelinating disorder predominantly affecting children. The disorder is characterized by encephalopathy, typically with additional neurological features (e.g., ataxia, cranial neuropathies, optic neuritis), and evidence of multifocal demyelination on neuroimaging. ADEM is classically considered a monophasic illness, though other relapsing demyelinating disorders may initially present with an ADEM phenotype.66,67 As children commonly present with antecedent infection prior to signs of neurological dysfunction, it is believed that ADEM is a post-infectious autoimmune disorder. Several pathogens have been associated with the disorder, with reports of ADEM-like illness during measles outbreaks dating back to the 18th century. 66 Antibodies targeting myelin oligodendrocyte glycoprotein are found in up to 60% of children with ADEM, and are frequently associated with relapses in other areas of the nervous system.68,69 To date, the role of infections in the development of these antibody-associated demyelinating disorders is poorly understood.
Narcolepsy
Beginning in the 1990s, researchers have suspected that certain forms of narcolepsy are caused by an autoimmune process. Narcolepsy type 1 (NT1), previously termed narcolepsy with cataplexy, was found to have a strong association with the human leukocyte antigen (HLA) DQB1*06:02, which was present in approximately 90% of this subgroup of narcolepsy patients. 70 Subsequently, the discovery of autoreactive CD4+ T cells leading to selective loss of hypocretin/orexin-producing neurons in the hypothalamus highlighted this as a possible mechanism for developing narcolepsy. 71 Additional support for the role of T cells in the disease include narcolepsy patients having polymorphisms in the T-cell receptor alpha locus. 72 In contrast, humoral mechanisms for narcolepsy have also be proposed with several groups reporting that individuals with narcolepsy have antibodies against Tribbles homolog 2 (anti-TRIB2) – a protein enriched in hypocretin neurons.73,74 Seropositivity was observed most frequently in narcolepsy with cataplexy near onset of disease, but rarely found in those without cataplexy and in those with more chronic disease.
Epidemiological studies have demonstrated that the incidence of narcolepsy fluctuates with seasonal infections such as influenza and strep throat. In 2009–2010, a striking increase in narcolepsy type I cases was seen in European countries, coinciding with a vaccination campaign against pandemic H1N1 Influenza A. The highest risk groups consisted of DQB1*0602 children and adolescents who had received the Pandemrix version of the H1N1 influenza vaccine. In 2010, increased narcolepsy cases in China were observed to peak about 6 months following winter H1N1 infections in a large retrospective study of patients with narcolepsy 75 ; 86% of the cohort was children and only 5.6% of the cohort recalled receiving a H1N1 vaccination.
Neurological syndromes in the setting of recent epidemics
Zika virus
Zika Virus was initially discovered in Uganda in the 1940s. In the 21st century, Zika virus managed to spread to the Pacific Islands in 2007, eventually reaching several countries in Central and South America starting in 2013.76,77 Prior to 2007, reports of human infection with Zika virus were reported only rarely, but its recent dissemination has seen outbreaks of a febrile illness with macular rash, arthralgias, and conjunctivitis. 78 Accompanying these outbreaks was an increase in GBS cases, which had an incidence up to 5.6 per 100,000.77,79,80 Initial reports from an outbreak in French Polynesia identified high rates of AMAN in post-Zika patients, though ganglioside antibodies were only identified in less than 50% of cases. 79 Among subsequent outbreaks in other nations, AIDP has been the most common GBS phenotype reported, with other GBS variants reported anecdotally. 81 Clinically, Zika-associated GBS tends to reach nadir much more rapidly than typical GBS (median 6 days), have shorter plateau, and present with more frequent facial weakness. 79 Other possible post-infectious presentations, including myelitis, meningoencephalitis, and chronic inflammatory demyelinating polyneuropathy, have also been reported in patients with confirmed or suspected Zika infections. 81
West Nile virus
West Nile Virus (WNV) is another important arboviral infection, responsible for epidemics in Europe and North America, starting in the late 20th century. While the majority of infections are asymptomatic, approximately 20% of individuals develop a mild febrile illness (West Nile Fever). 77 Approximately 1 in 150 infected individuals develop neuroinvasive WNV infections. Clinical manifestations of neuroinvasive WNV include aseptic meningitis, encephalitis (typically with prominent extrapyramidal symptoms), or a gray matter-centric myelitis with flaccid paralysis.77,82,83 Immune-mediated neurological complications have been reported in patients with confirmed WNV infection, suggesting the virus can trigger loss of tolerance. Leis and colleagues reported a series of patients that developed acetylcholine receptor antibody positive MG in the months after neuroinvasive WNV infection. 84 Further evidence of a possible association comes from a small, single-center study that identified serologic evidence of previous WNV infection in 17% of patients with MG, all of whom were asymptomatic. 85 Larger studies are needed to confirm an association between MG and WNV. Other neuromuscular presentations reported post-WNV include GBS, brachial plexopathies, multifocal motor neuropathy, and myositis.86–88 Among CNS disorders, a case of autoimmune encephalitis with glycine antibodies and a case of stiff person syndrome have also been reported.89,90
Chikungunya
Chikungunya is a mosquito-borne alphavirus that causes a systemic illness with myalgia, headache, fever, and debilitating arthralgia. The virus became a major public health concern starting in 2004, when a new strain began to spread across island nations in Asia, then to the Americas, infecting millions. 91 Neurological sequelae of chikungunya infection are increasingly recognized, and are strongly associated with the need for intensive care. 92 Encephalopathy is the most common neurological symptom reported, with some publications providing supporting evidence of encephalitis in subjects with chikungunya infection. 93 Spinal cord syndromes with evidence of myelitis have also been frequently reported, either in isolation or in conjunction with other manifestations. Cases of acute neuropathic presentations consistent with GBS are also recognized.94,95 Evidence supports the possibility of neurotropism for the virus, though the description of ADEM and GBS raise the possibility of post-infectious mechanisms for certain chikungunya-associated presentations.
Coronaviruses
Coronaviruses have emerged as an important pathogen in humans in the 21st century, with two novel coronaviruses [severe acute respiratory syndrome coronavirus 1 (SARS-CoV-1) and Middle East Respiratory Syndrome (MERS)], resulting in epidemics of pulmonary disease with high mortality. 96 During outbreaks of these viruses, potential neurological manifestations were reported. Seizures, rhabdomyolysis, stroke, and acute neuropathy were reported in patients with SARS-CoV-1, with possible neurotropism suggested by the detection of viral RNA in CSF and tissue specimens.97–102 Similarly, diverse manifestations (mostly encephalopathy and seizures) were reported in patients with MERS, though evidence of neurotropism has not been reported to date.102–105
Recently, a novel coronavirus (SARS-CoV-2) causing the respiratory illness Coronavirus Disease -2019 (COVID-19) was discovered in the Hubei province in China. The virus quickly spread globally and was declared a pandemic by the World Health Organization on 11 March 2020. 106 To date, there have been over 10.5 million confirmed COVID-19 cases worldwide, with over 500,000 deaths. 107 Diverse neurological complications are being reported in the literature, though pathophysiologic mechanisms are still poorly understood. Manifestations such as encephalopathy or cerebrovascular complications may be due to dysregulated homeostatic functions or a coagulopathy, rather than direct neurotropism or targeted inflammatory response. 108 However, reports of neurological symptoms weeks after infection suggest a subset of neurological presentations result from post-infectious autoimmunity.
Central and peripheral nervous system complications of COVID-19 infection have been reported. Anosmia and dysgeusia are the most common neurological symptom, recognized in up to 60% of hospitalized COVID-19 patients. 109 The pathophysiologic mechanisms for anosmia are unclear, but in a case report demonstrating neuroimaging abnormalities within the posterior gyrus rectus, direct viral invasion of the olfactory cortex was postulated. 110 Cases of GBS associated with COVID-19 are also emerging in the literature, with 40 cases reported to date. Reported phenotypes have predominantly included demyelinating polyneuropathies, though cases of Miller Fisher syndrome and axonal neuropathies has been reported.39,111–132 Molecular mimicry is suspected in association with SARS-CoV-2 associated GBS, given interactions between the spike protein and cell-surface gangliosides, and the presence of antiganglioside antibodies in reported cases. 37
Possible cases of meningoencephalitis, with neuropsychiatric features, altered mental status, and seizures, have been reported in hospitalized COVID-19 patients. While case descriptions are highly variable, some reports provide evidence of inflammation on neuroimaging and CSF analysis, fulfilling criteria for encephalitis.133,134 Few reports have identified SARS-CoV-2 RNA in the CSF, and a single post-mortem study identified the virus in neural tissue on electron microscopy, suggesting these cases may be due to direct viral invasion of the CNS.135–137 The virus may be capable of triggering antibody-mediated encephalitis, as suggested in a recently reported case of NMDA-R encephalitis in a COVID-19 patient hospitalized with acute psychiatric disturbance and hypoxia. 138 Other CNS manifestations, including ADEM and an acute myelitis, have been reported in COVID-19 patients.139,140
Post-vaccination neurological syndromes
Acute disseminated encephalomyelitis
The first reports of neurological syndromes being temporally associated with immunizations occurred in the 19th century following the introductions of smallpox (cowpox) and rabies vaccines.141,142 Initially, post-vaccination ADEM was thought to be caused by the vaccine’s viral components, but later studies suggest that contamination with CNS tissue, particularly in the case of the rabies vaccine, was felt to be the antigenic trigger. This hypothesis is substantiated by reduced incidence of post-vaccination ADEM following changes in vaccine development that utilized recombinant proteins, rather than in vivo infected animal tissue.
Numerous other vaccines have been implicated including diphtheria–tetanus–polio, measles, mumps, rubella, Japanese B encephalitis, pertussis, influenza, and hepatitis B. 143 In actuality, post-vaccination encephalomyelitis is extremely rare, with an incidence of 0.1–0.2 per 100,000 vaccinated individuals and accounts for less than 5% of ADEM cases. Past studies suggest that ADEM is significantly more likely to occur following infection rather than immunization, with an incidence of ADEM as high as 1 in 1000 cases of measles virus infection versus 1–2 million per live measles vaccine. 141
Guillain–Barre syndrome
In 1976, the incidence of GBS increased to 1 in 100,000 in the weeks following the release of an H1N1 influenza A vaccination. 144 The stark increase resulted in a recall of the vaccine, and raised concern about the role of influenza immunization in the pathogenesis of GBS. Intensive epidemiologic study of GBS with subsequent influenza vaccines (including a subsequent H1N1 vaccination in 2009) have demonstrated a small increases in incidence with vaccination,22,145 with some studies have demonstrated a protective effect of flu vaccination for preventing GBS. 146 Furthermore, evidence suggests patients with a history of GBS do not experience recurrence following influenza vaccination. 147
Diagnostic and treatment considerations
Initial evaluation
The initial evaluation and treatment of a suspected post-infectious neurological disorder mirrors that of other neuroimmunological disorders. Presentations with severe or progressive symptoms require close monitoring, and intensive care may be necessary for refractory seizures, autonomic dysfunction, and or signs of increased intracranial pressure.148,149 Neuroimaging and CSF analysis are important parts of the diagnostic evaluation as they help identify evidence of inflammation and rule out alternate etiologies. Cancer screening is commonly recommended in many antibody-associated disorders given the possibility of a paraneoplastic etiology. 150 While many of the disorders in this article are not commonly associated with a neoplasm, anti-NMDA-R encephalitis has an association with ovarian teratoma, typically in young women with the disorder. 151 Screening should be considered on a case-by-case basis, based primarily upon risk factors and clinical context.
Antibody testing
Many autoimmune disorders discussed in this review are associated with antibodies targeting glial or neuronal antigens. Testing is available through several commercial laboratories. As different autoantibodies can be associated with overlapping clinical presentations, it is generally advised to send a “panel” of antibodies. 152 Presentations with classic demyelinating features (optic neuritis, transverse myelitis) should strongly consider testing for aquaporin-4 and MOG antibodies. Patients with clinical features consistent with a GBS variant, particularly AMAN or Miller Fisher syndrome, should undergo testing for ganglioside antibodies. Several antibodies targeting cell-surface proteins or intracellular antigens have been associated with autoimmune encephalitis (please see the review on Autoimmune Encephalitis in this edition for details). In the evaluation for autoimmune encephalitis, it is generally recommended that samples from both serum and CSF should be submitted for testing, as detection for certain antibodies (e.g., NMDA-R) is more sensitive in CSF. 151
The presence of an autoantibody on testing may indicate an autoimmune disorder, but it is important to consider the clinical context when interpreting results. Low titers of certain antibodies may have limited clinical significance; for example, low titers of GAD65 antibodies can be present in the general population and in individuals with type 1 diabetes mellitus.153,154 Commercial laboratories may also report values for voltage-gated potassium channel antibodies, though the importance of this antibody in the absence of LGI1 or Caspr2 antibodies has been called into question. 155 Diagnostic criteria for many autoimmune disorders incorporate clinical imaging and CSF findings to facilitate early treatment in high probability cases, or in instances where antibody testing is unavailable.152,156 Antibody negative presentations of disorders such as autoimmune encephalitis are increasingly reported in the literature, highlighting the importance of clinical context in decision making for autoimmune neurological disorders. 157
Treatment considerations
While disorders such as SC may be managed with symptomatic therapies in milder cases, the treatment of most neuroimmunological disorders relies on the use of immunomodulating or immunosuppressive measures. Developing an immunotherapy plan for a patient is multifaceted, taking into account patient comorbidities, clinical phenotype and presumed pathophysiology of the neuroimmunological disorder. Monophasic disorders such as GBS may only require one-time intervention with immunotherapy, whereas disorders with high risk of relapse may require further immunosuppressive agents. Clinical trial data is lacking for many of the antibody-mediated CNS disorders, and experience from other neurological or systemic autoimmune disorders is often utilized to guide management.150,158,159
Guillain–Barre syndrome
Strong evidence suggests immunotherapy plays an important role in the management of GBS. IVIG and plasmapheresis have both demonstrated efficacy in trials at improving outcomes at 6 months, with comparative studies of the two interventions finding similar efficacy.160–163 Furthermore, the administration of IVIG following a course of plasmapheresis did not confer any additional benefit. 164 Studies assessing the role of corticosteroids failed to demonstrate improvement in GBS patients, with one small study of oral steroids suggesting deleterious effects. 165 Other treatments, including interferon beta and eculizumab, have been studied in small trials of GBS patients, but larger studies are needed to investigate their role in management. 166 After initial treatment, approximately 10% of patient with GBS will experience worsening of symptoms, typically within 8 weeks of initial presentation. A second round of treatment is often considered in these instances. In patients with a relapse several months from the initial presentation, or with a reported slower progression atypical for GBS, a broadened differential diagnosis and treatment approach should be strongly considered.21,167
Autoimmune encephalitis
The decision to initiate immunotherapy in suspected post-infectious autoimmune encephalitis can be challenging. When faced with a patient with recent HSE presenting with new neurological symptoms, clinicians may hesitate to start immunosuppressive treatment due to concerns that it may blunt the antimicrobial immune response or promote viral replication. Experimental models suggest that treatment with glucocorticoids does not exacerbate active herpes simplex infection.168,169 Furthermore, a recent systematic review determined that, among 43 post-HSE cases, no patients had experienced recurrence of herpes simplex following treatment for autoimmune encephalitis, despite the use of first and second-line immunotherapy. 168 Clinical features may also serve to guide treatment decisions, as features like psychosis and hyperkinetic movements are more frequent in NMDA-R encephalitis than in HSE. 44 This constellation of evidence has led authors to conclude that, after an expeditious evaluation for recurrence in the setting of HSE, the treatment of post-infectious autoimmune encephalitis should mirror that of cases without a known infectious trigger.168,170
High dose corticosteroids, intravenous immunoglobulin (IVIG), and plasmapheresis have all been utilized as first-line agents for autoimmune encephalitis.150,171 Treatment response is generally assessed based upon clinical improvement in symptoms, though repeat imaging or spinal fluid analysis may be useful in certain instances. In cases of NMDA-R encephalitis that fail to improve with first-line immunotherapy, second-line therapy (typically with cyclophosphamide or rituximab) have demonstrated improvement long-term functional outcomes. Evidence also suggests rituximab may reduce the risk of clinical relapse. 151 Tocilizumab and bortezomib have also been reported to be beneficial in refractory cases.172,173
Conclusion
Infections represent an important risk factor for the development of neuroimmunological disorders. Certain autoimmune neurological disorders have a close association with specific infectious pathogens, which likely arise through diverse immunological mechanisms. A post-infectious autoimmune disorder is an important consideration in the setting of new neurological symptoms temporally associated with infectious symptoms. Prompt recognition and treatment of autoimmune neurological disorders can lead to favorable outcomes.