
Abstract
Extensive research over the last decades basically failed to identify a common cause of noninfectious inflammatory central nervous system (CNS) demyelinating disease. To a great extent, this may reflect that the group of inflammatory CNS demyelinating disorders likely contains multiple pathogenetically distinct disease entities. Indeed, the greatest success so far in deciphering the pathogenesis of a CNS demyelinating disorder resulted from the discovery of anti-aquaporin (AQP)-4 antibodies (ab), which allowed progressive delineation of neuromyelitis optica (NMO), formerly considered a variant of the most common CNS demyelinating disorder, multiple sclerosis (MS), as a distinct disease. Nowadays, AQP-4+ NMO is considered an autoimmune astrocytopathy, in which CNS demyelination occurs only as a consequence of a primary destruction of astrocytes. Delineating these patients concomitantly revealed that not all patients presenting with clinically NMO-suggestive disease phenotype express AQP-4 ab, which created the pathogenetically undefined category of NMO spectrum disorders (NMOSD). Recent investigations discovered that a subgroup of these AQP-4– NMOSD patients produce an ab response against myelin oligodendrocyte glycoprotein (MOG), a molecule expressed on the outer lamella of the myelin sheath. Using pathophysiologically meaningful cell-based assays, this humoral response is extremely rare in adult MS and absent in classical AQP-4+ NMO, sharply differentiating the evolving group from both established disorders. In this review, we summarize available clinical, immunological and histopathological data on patients with MOG+ CNS demyelinating disease. By comparing this clearly distinct cohort to AQP-4+ NMO as well as MS, we propose that MOG+ CNS demyelinating disease represents a distinct novel disease entity. In addition to its diagnostic value, we furthermore provide mechanistic insight on how this peripheral anti-MOG ab response may be of pathogenetic relevance in triggering acute flares of inflammatory CNS demyelination.
Keywords
Introduction
B cells, plasma cells and plasma cell secreted antibodies (ab) may play an important role in the development of central nervous system (CNS) demyelinating disorders, a notion that was significantly revived by the fulminant success of anti-CD20-mediated B-cell depletion in recent clinical trials.1–3 In multiple sclerosis (MS), intrathecal immunoglobulin (Ig) production by clonally expanded and locally supported plasma cells remains a hallmark diagnostic finding, and ab depositions along with complement activation can be found in areas of active CNS demyelination. 4 Despite exhaustive investigations, no common autoantigen has been identified so far, 5 likely relating to the fact that MS is a heterogeneous disorder and may comprise several disease entities. 6 Based on the leading pathology of demyelination, possible autoantigens have been primarily projected into the myelin sheath and the oligodendrocyte. Here, myelin oligodendrocyte glycoprotein (MOG) exposed on its outer most lamella is a prime candidate. Since it is not expressed in the thymus and in peripheral organs it is thought that mechanisms to ensure immunological tolerance against MOG are less well established compared with other CNS antigens. 7 Peripheral injection of MOG can induce several experimental, primarily T-cell-mediated models of MS in a wide range of species. Using an adequate immunization regimen, a disease-consolidating humoral response against MOG can be raised. 8 Accordingly, adaptive immune responses against MOG have been widely investigated in patients with MS and related demyelinating disorders. These studies however often generated nonconclusive and internally conflicting data, substantially adding to the complexity of the subject. In this regard, early enzyme linked immunosorbent assay (ELISA)-based investigations suggested that ab response against MOG may occur frequently in patients with MS, and that the humoral response against this and other myelin antigens may predict disease severity. 9 Methodological improvements towards cellular expression of MOG and accordingly its recognition within a biosimilar context basically zeroed this association.10–12 The respective studies revealed that anti-MOG ab determined by functionally meaningful cell-based assays are rare in adult MS, while they define a subgroup of paediatric patients with acquired CNS demyelination. Their highest titres were found in cases of acute disseminated encephalomyelitis (ADEM). 13 The majority of these children however showed a rapid decline of anti-MOG ab after recovery from the monophasic clinical episode, raising the question whether MOG-reactive ab are causative in these cases of paediatric ADEM or whether they may occur secondarily as a consequence of massive myelin degradation. Regardless, these findings proved the principle that peripheral anti-MOG ab can be found when parts of the CNS undergo inflammatory demyelination, and accordingly, that MOG is sufficiently immunogenic in humans.
In this review, we summarize and discuss entirely novel or currently emerging data suggesting that while the general association with MS can be assumed as negative, a small subgroup of adult individuals with CNS demyelination contains serum anti-MOG ab. These patients present with internally homogenous clinical, radiological and immunological features jointly distinct from both MS and neuromyelitis optica (NMO). We propose that anti-MOG ab may thus allow delineation of these patients from the ‘core disease MS’, shaping a novel disease entity. We assume that after giant discoveries and corresponding setbacks, these latest developments will bring the twisting and winding path of MOG-related research in CNS demyelinating disorders to a new beginning.
The evolvement of NMO as an autoimmune astrocytopathy
The driving force behind understanding NMO was the conception of it being different from MS. This development started by a careful and vigilant description of its clinical and later radiological features. 14 In 1894, Eugene Devic described a novel syndrome characterized by acute myelitis and optic neuritis which he accordingly termed ‘neuro-myélite optique aiguë’. Together with his student Fernand Gault he subsequently defined clinical and early pathological properties of the disease, which were summarized in Gault’s thesis. 14 These crucial steps provided the basis for the later observations that besides the preferential affection of the optic nerve and spinal cord, acute attacks of NMO were more severe, longer lasting and relatively persisting compared with relapsing–remitting (RR)-MS. 15 On the contrary, NMO appeared not to transition into a secondary progressive course, as commonly observed in MS. 16 Subsequently, these clinical features were recognized to be reflected by the relative absence of magnetic resonance imaging (MRI)-detectable focal inflammation in the brain and the predominant occurrence of longitudinally extensive spinal cord lesions. 17 NMO was further found to peak at a later stage in life 15 and to have an even stronger female predilection than MS. 18 Lastly, certain MS medications like interferons or natalizumab essentially failed or even worsened its poor prognosis,19–21 suggesting early on that NMO was not a severe variant of MS, but a distinct disease entity with a pathogenesis different from MS.
The next pivotal step in this direction was the identification of ab against aquaporin (AQP)-4 in NMO cases, 22 which is probably the biggest discovery in CNS demyelinating disorders thus far. AQP-4 is a water channel that is widely expressed, for example, in the kidney. 23 Within the CNS, AQP-4 is primarily found at the end feet of astrocytes, 24 most densely expressed in the optic nerve and the spinal cord, where they are in close contact with oligodendrocytes. 25 Of note, AQP-4 is not expressed on oligodendrocytes themselves, the cells responsible for CNS myelination. 24 While the pathophysiological role of anti-AQP-4 ab was unclear at the time of discovery, 26 they rapidly evolved into a prime diagnostic tool. 27 This novel marker now flanked, but also in part challenged the former clinical and radiological diagnostic criteria of NMO, as for instance AQP-4 ab could be found in cases of assumed MS, probably misdiagnosed due to the presence of inflammatory brain lesions. 28 The predominant accurateness of AQP-4 ab for the diagnosis of NMO and accordingly the necessity to refine the ‘radiological dogma’ that NMO exclusively affects optic nerves and spinal cord was brought forward by the understanding that AQP-4 ab are directly pathogenic in NMO. 29 First, it was observed that the presence of AQP-4 ab and in particular their capability to destroy membrane-bound AQP-4 in vitro correlated with disease activity and severity.30,31 Second, comparative histopathological analyses of NMO lesions 32 revealed homogenously an extensive devastation of astrocytes in areas of active inflammation associated with perivascular ab and complement deposition and loss of astrocytic AQP-4. 33 Importantly, demyelination was determined to occur only as a consequence of this process.34,35 These findings firmly established that NMO is an independent disorder and promoted its recognition as an autoimmune astrocytopathy, 36 mediated at least in part by anti-AQP-4 ab. This pathophysiological concept of NMO explains several of its key features, such as the continuous expansion of lesions as well as the poor prognosis of NMO attacks; by far most importantly, deciphering the pathogenesis of NMO pioneered and evolved targeting B cells and the humoral response against AQP-4 as the gold standard in the treatment of NMO. Notwithstanding these giant steps in understanding NMO, several questions remain: first, why are AQP-4 ab selectively destroying astrocytes despite the almost ubiquitous expression of AQP-4? 37 Second and possibly related, why and how are these CNS-directed ab generated in the periphery, yet rarely found in the cerebrospinal fluid (CSF) 38 and by which mechanism do they cross the blood brain barrier?
MOG antibody associated CNS demyelination
The specificity of anti-AQP-4 ab for the diagnosis of NMO is extremely high. 39 With methodological improvements, their initial sensitivity of around 70% continuously increased over the last years, ultimately questioning whether the now sharply defined diagnosis of NMO exists without the pathogenic contribution of anti-AQP-4 ab. 40 This development collaterally generated a subgroup of patients formerly diagnosed as having NMO by clinical and radiological criteria, in whom despite extensive testing no anti-AQP-4 ab could be found. On the search for a fitting diagnosis other than classical AQP-4+ NMO, but also distinct from MS, the overarching category of NMO spectrum disorders (SDs) was generated, 41 further divided into AQP-4+ and AQP-4– NMOSD. The evolving perception of classical NMO as an AQP-4 ab-mediated astrocytopathy, however, raises the question whether AQP-4– CNS demyelination can truly resemble NMO in a pathophysiological manner, and accordingly, to what extent the category of AQP-4– NMOSD is useful. 42 Based on this dilemma, it raised enormous interest that in about a third of these patients ab against MOG could be identified.43–47 In the following sections we try to make the case that by clinical, radiological, histological and immunological criteria this internally homogenous patient cohort is different from both NMO and MS, and that accordingly, anti-MOG ab associated CNS demyelination will most likely evolve as a distinct disease entity.
Subgroups of patients expressing MOG antibodies
The distinction of a MOG ab+ subgroup of patients with demyelinating CNS disease was only possible when it was discovered that a cell-based assay is needed to detect disease-associated MOG ab. Before, several ELISA and western blot assays yielded conflicting results. Neither a disease-specific occurrence of MOG ab nor a prognostic value of these ab could reproducibly be shown. 48 The seminal work by O’Connor established that in vitro translated MOG tetramers were able to detect MOG ab in a significant subgroup of paediatric patients with CNS demyelination. 11 This initial report was followed by many other groups that could largely reproduce their findings.13,44,49–52 The epitope specificity of MOG ab in the paediatric population has been analysed using different MOG mutants. 53 Seven different epitope patterns could be identified. A longitudinal follow up of these patients indicated that the epitope was stable over time with no indication of an epitope spreading within the molecule. Only few of these patients had MOG ab that cross reacted with rodent myelin. While one study indicated a larger proportion of patients with MOG ab cross reacting with rodent tissue and suggested that this feature of cross reactivity indicates the clinical phenotype of NMOSD, 46 another recent report suggests that only a subset of human MOG ab are reactive to mouse or rat. 54 Only few reports argued that an ab response against conformational MOG can really be detected in a larger group of adult patients with MS.55,56 It is currently accepted that adult patients with MS in general do not mount a humoral immune response against MOG. However, there is increasing evidence that an ab response against MOG defines a small subgroup of patients with distinct clinical features that predominantly manifest with concomitant severe brainstem and spinal cord involvement, have a severe disease course with high relapse rates, and do not respond to several disease-modifying MS therapies. 57 In addition there is another subgroup of adult patients with ab against the glycine receptor a1 subunit (GlyR) or the N-methyl-D-aspartate receptor (NMDA-R) that also have MOG ab.58,59 The clinical features of these two subgroups were different: patients with GlyR ab had recurrent isolated optic neuritis whereas the patients with NMDA-R ab had independent episodes of NMOSD, brainstem or multifocal demyelinating syndromes.
In the paediatric population, MOG ab were mainly detected in monophasic ADEM. 12 However, persisting titres of MOG ab were also reported in children with recurrent demyelinating syndromes. In this patient group, MOG ab tended to correlate with the occurrence of relapses, whereas in monophasic cases, MOG ab disappeared after resolution of clinical symptoms.13,60 Since these paediatric patients were shown to harbour MOG ab it was speculated that these ab might be present only in the very early stages of adult MS. However, a study investigating the presence of MOG ab in this group of patients did not find evidence for this hypothesis. 10
MOG ab seem to be specific for autoimmune demyelination because they could be used to separate viral encephalitis from demyelinating autoimmune encephalopathy. 61 In adults, MOG ab are mainly detected in patients with a clinical phenotype that resembles NMO rather than MS but also has features that distinguishes this subgroup from AQP-4 ab+ NMO.43–45,62–69
Clinical presentation, radiological and laboratory findings in adult patients who are MOG ab+
Unlike paediatric ADEM with MOG ab which often manifests after a preceding infection, adult CNS demyelination associated with MOG ab is normally not linked to an infection or vaccination. However, there are a few case reports of infection or vaccination associated demyelinating CNS syndromes in which ab against MOG could be detected that is reminiscent of the paediatric ADEM population.70–72 MOG ab seem to occur largely independent of other autoimmune disorders while AQP-4 ab were also found in other autoimmune diseases with involvement of the CNS. 72 A large cohort study detected MOG ab in 19.6% of patients with myelitis or optic neuritis. 73 While individual titres of MOG ab in the serum can fluctuate over time and might correlate with disease activity and treatment, their overall detection persisted over time.73,74 Patients with MOG ab are on average younger than patients with NMO and AQP-4 ab46,72,75 with a gender ratio of 2–3:1 (female:male), affecting on average more male patients than classical NMO (Table 1). Several studies showed that there is no overlap between patients who are MOG and AQP-4 ab+, indicating two clinically and pathologically different disease entities. MOG ab were most commonly found in patients with isolated optic neuritis 76 and combined optic neuritis/myelitis and only occasionally in isolated myelitis. Similar to AQP-4+ NMO, MOG ab are mainly found in serum and there is no intrathecal production of MOG ab. 73 However, an increased pleocytosis including some cases with increased neutrophils in the CSF was noted in different reports.65,73 These CSF changes were accompanied by increased cytokines or chemokines that are related to B cells and neutrophils. 77 Interestingly, an unspecific activation of B cells that is associated with MS and leads to an intrathecal antibody response against different viruses could not be shown in MOG ab associated CNS disease. Along the same lines, oligoclonal bands in the CSF were usually negative in patients who were MOG ab+. This distinct set of features is probably of greatest value in differentiating characteristics for clinical diagnosis38,78,79 (Figure 1).
Clinical and magnetic resonance imaging (MRI) characteristics of patients with myelin oligodendrocyte glycoprotein (MOG) antibody (ab) associated central nervous system demyelination (MOG ab), aquaporin 4 (AQP-4) ab+ neuromyelitis optica (NMO) or multiple sclerosis (MS).
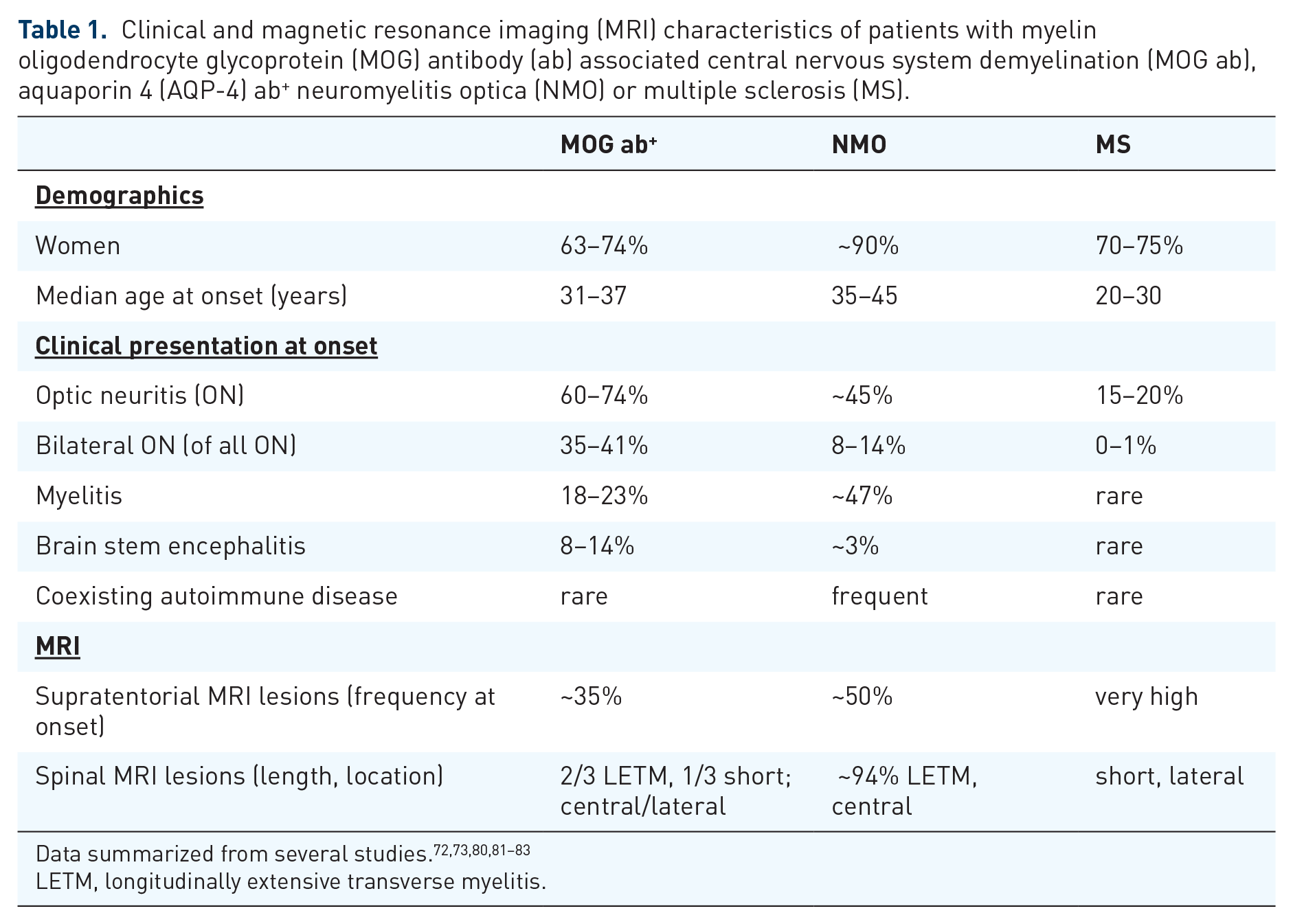
LETM, longitudinally extensive transverse myelitis.

Laboratory findings in central nervous system (CNS) demyelinating disorders. Signs of B-cell activation in multiple sclerosis (MS) occur predominantly within the CNS, whereas both neuromyelitis optica (NMO) and myelin oligodendrocyte glycoprotein (MOG) antibody (ab) associated CNS demyelination are characterized by CNS-directed ab produced in the periphery. Data summarized from several studies.72,73,78,81,82,84,85 AQP-4, aquaporin 4; MRZ, measles, rubella, varicella zoster virus.
MRI imaging can also be used to separate MOG ab associated CNS disease from AQP-4 ab+ NMO and MS. 86 The localization of lesions in MOG ab disease is often confined to the pons and adjacent to the fourth ventricle. Brainstem lesions in MOG ab disease can have a fluffy appearance. In contrast, ovoid lesions adjacent to the lateral ventricles, Dawson’s fingers, and T1 hypointense lesions were rather specific for MS and less frequently seen in NMO and MOG ab associated disease. Whereas a discrimination between MS and MOG/AQP-4 associated diseases was possible, the dissociation of both ab associated CNS diseases by MRI was less accurate. 87
The clinical presentation of patients who are MOG ab+ is characterized by a more frequent involvement of optic nerves than spinal cord.63,88 Also a simultaneous affection of both optic nerves seems to be a hallmark of patients who are MOG ab+ compared with NMO or MS. 66 Of interest, in patients with a clinically unilateral optic neuritis, the nonaffected eye often shows a subclinical atrophy, suggesting that the involvement of the optic nerve appears to be a clinical sign in patients who are MOG ab+. 89 There might be anatomic distinction with regard to the location of the inflammation of the optic nerve between MOG- and AQP-4-associated optic neuritis: MOG-associated neuritis predominantly affects the retrobulbar region whereas AQP-4-associated optic neuritis is located intracranially. A subgroup of patients with MOG ab associated optic neuritis showed contrast enhancement in the surrounding fat tissue of the optic nerve. 72 Investigations of the lesion location in the spinal cord revealed that the lower part of the spinal cord might be preferentially affected in patients who are MOG ab+.62,68 Lastly, compared with patients with AQP-4+ NMO, patients who are MOG ab+ appear to present more frequently with seizures. 90
Characteristics of brain and spinal cord MRI were further analysed in another cohort of 50 adult patients with MOG ab. 80 In this cohort infratentorial lesions in the brainstem and cerebellum were quite common, with an incidence of around 30%. However, supratentorial lesions were also found in approximately half of the patients. Spinal cord involvement was seen in two thirds of the patients, with a majority of these patients showing longitudinal extensive lesions. Twenty-seven percent of patients with brain lesions fulfilled the Barkhof MRI criteria for MS diagnosis. The MRI findings in general substantiate that MOG ab associated disease shares clinical as well as imaging features with both neighbouring diseases MS and NMO. A MRI-based differentiation is probably only possible between MOG ab associated CNS disease and MS. AQP-4 and MOG ab associated diseases can be very similar in their MRI characteristics.
Due to the paucity of patients who are MOG ab+ and a rather short follow up, the disease prognosis is still controversial. Some studies reported a more benign disease course with fewer relapses in patients with MOG ab.65,91 It has therefore been speculated that due to the lower relapse frequency and a better functional recovery, patients who are MOG ab+ have a better prognosis.13,45,46,58,62,64,67 A recent German cohort study indicated a multiphasic course in 80% and an annualized relapse rate of 0.9. 72 One third of patients with optic neuritis and approximately half of patients with myelitis made a full recovery in this cohort. The rest of the patients showed residual deficits after the relapses. Two other studies demonstrated, however, that the retinal neuro-axonal damage measured after an acute optic neuritis in patients who were MOG ab+ was as severe as in patients with AQP-4 ab+ NMO, suggesting that an equal deficit can remain from individual relapses in patients who are MOG ab+.92–94 Due to the small number of cases, the therapeutic response of MOG ab associated CNS diseases is not well known. Most patients are treated with classical immunosuppressants like azathioprine. In refractory or more aggressive cases, B-cell depleting antibodies such as rituximab are also used. 95 Similar to classical NMO, immunomodulation with interferons might have a negative impact on disease activity. 72
Histopathological findings in adult patients who are MOG ab+
Histopathological descriptions of CNS lesions from patients with serum MOG ab are rare. A systematic literature search identified a total of seven brain biopsies from six patients who had histological and immunohistochemical work up. All biopsies were from adult patients who presented with a variety of clinical symptoms and MRI findings, including tumefactive MS-like lesions,96,97 an NMOSD-like phenotype57,98 or an ADEM-like presentation. 99 In all six patients, the presence of MOG ab was confirmed by cell-based assays and brain biopsies were performed for differential diagnostic reasons. All cases showed a plaque-like myelin loss with relative axonal preservation. Inflammatory infiltrates, consisting mainly of T cells and macrophages or microglia, were present perivascularly or within the brain parenchyma. Demyelination was confluent and not restricted to the perivascular spaces as it is typically seen in ADEM patients, thus excluding this differential diagnosis pathologically. NMO lesions are characterized pathologically by an astrocytopathy with loss of AQP-4 expression and secondary demyelination with oligodendrocyte loss (Figure 2). Pathogenetic mechanisms in NMO involve peripheral anti-AQP-4 antibodies targeting astrocytes. Pathologically, perivascular antibody and complement deposits as well as presence of eosinophilic granulocytes are typically seen in acute NMO lesions. None of these NMO-specific pathological features was seen in the six lesions from patients who were MOG ab+. Eosinophils were not detected; astrocytes and AQP-4 were preserved, as were oligodendrocytes in the lesions, making the diagnosis of NMO highly unlikely in these patients. All seven biopsies from the six patients who were MOG ab+ revealed actively demyelinating lesions pathologically. In five out of the six cases, activated complement deposits (C9neo complex) were seen in macrophages and along myelin sheaths in areas of active demyelination. In addition, in all but one case, oligodendrocytes and their precursors were still present within the lesions. These features are clearly compatible with MS pattern II lesions (Figure 2, Table 2).100–103 In conclusion, these six MOG ab+ cases teach us that MOG ab+ demyelinating encephalomyelitis is an inflammatory demyelinating disease; it is pathologically neither ADEM nor NMO; and it shares many pathological features with MS, especially the antibody- or complement-mediated pattern II. Neither MOG protein nor oligodendrocytes are lost in lesions from patients who are MOG ab+, raising the question of the pathogenetic role of anti-MOG ab in these patients in contrast to the clear pathogenetic role of astrocyte-targeting anti-AQP-4 antibodies in NMO. Whether there may be an overlap between MOG and AQP-4 ab still remains open. Thus far, only one case is described in the literature, which shows an overlap of MOG and AQP-4 ab positivity. 104 In this patient, the cerebral lesions showed characteristic features of MS whereas the lesions in the optic chiasm resembled NMO with astrocytopathy, AQP-4 loss and perivascular complement deposits.
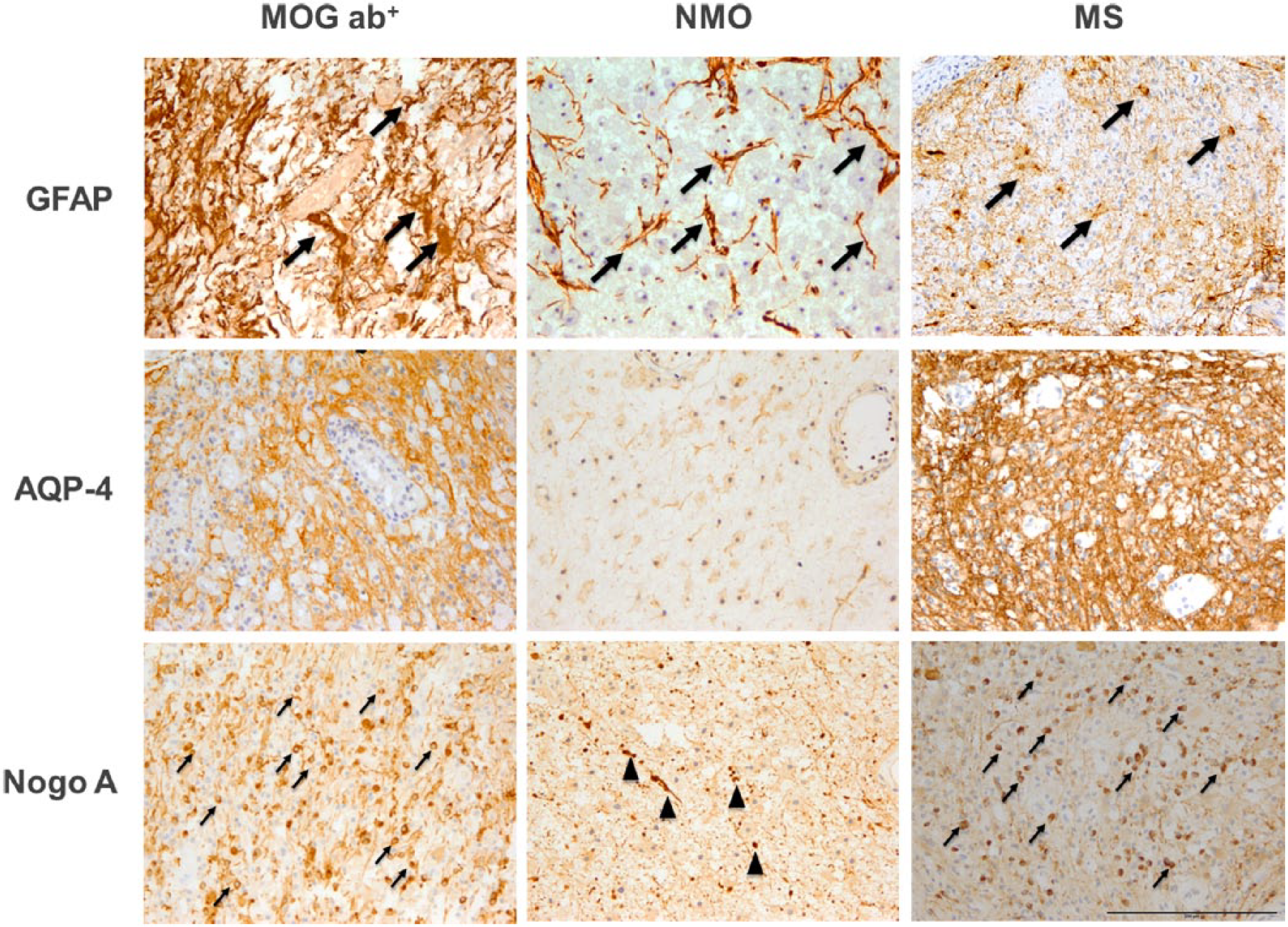
Representative histopathological findings in myelin oligodendrocyte glycoprotein (MOG) antibody (ab) associated central nervous system demyelination (MOG ab; left column, a, d, g), aquaporin 4 (AQP-4) ab+ neuromyelitis optica (NMO; middle column, b, e, h) and multiple sclerosis (MS; right column, c, f, i). Early active demyelinating lesions were stained for astrocytes/gliosis (glial fibrillary acidic protein; GFAP; upper panels a–c). AQP-4 (middle panels, d–f) and mature oligodendrocytes (Nogo A; lower panels g–i). NMO lesions show the characteristic dystrophic astrocytes (b, arrows) indicative of astrocytopathy whereas MOG ab (a) and MS (c) show reactive astrocytes (arrows) and a dense GFAP+ fibre network (upper panels). AQP-4 is preserved in MOG ab (d) and MS (f) whereas it is lost in NMO lesions (e) (middle panels). Mature oligodendrocytes are preserved in MOG ab (g) and MS (i) in contrast to NMO (h) where mature oligodendrocytes are significantly lost (lower panels) The Nogo A+ structures in (h) represent axonal spheroides (arrowheads) and not mature oligodendrocytes. Magnification: ×20 (scale bar 200 µm).
Histopathological findings in patients with myelin oligodendrocyte glycoprotein (MOG) antibody (ab) associated central nervous system demyelination (MOG ab), aquaporin 4 (AQP-4) antibody positive neuromyelitis optica (NMO) or multiple sclerosis (MS).

n. r., not reported.
Possible effector mechanisms of anti-MOG ab in CNS demyelinating disease
While MOG protein or oligodendrocytes are not entirely extinguished in CNS lesions of patients who are MOG ab+, MOG ab may nevertheless directly bind within the CNS and accelerate ongoing CNS demyelination. Supporting this concept, patients who are MOG ab+ show an isolated elevation of myelin basic protein indicative of primary CNS demyelination,105,106 while glial fibrillary acidic protein (GFAP), a marker of astrocyte destruction often found in classical NMO, is not increased in patients who are MOG ab+. Besides activation of complement, binding of anti-MOG ab to MOG within the myelin sheath may facilitate its recognition by antigen-presenting cells (APCs) within the CNS, accelerating tissue destruction by T cells. 107 Furthermore, MOG ab may hinder binding of potentially restoring nerve growth factor to MOG 108 and induce repartition of MOG into lipid rafts, perturbing oligodendrocyte physiology. 109
Besides such direct targeting of the CNS myelin sheath, MOG ab may exert additional properties outside of the CNS, a notion supported early on by the predominance of ab against MOG in the serum, while their intrathecal production appears to be extremely rare. Paralleling AQP-4+ NMO, this finding advances the central question why this ab response against CNS antigens is raised in peripheral compartments, and even more so how MOG which is exclusively expressed within the CNS is so dominantly recognized by the peripheral immune system. One possibility is that in ongoing inflammation CNS infiltrating immune cells recognize CNS MOG and export antigen recognition or MOG antigen itself by subsequently leaving the CNS towards deep cervical lymph nodes,110,111 where newly appreciated lymphatic vessels drain brain interstitial fluid.112,113 Traces of myelin have indeed been detected in deep cervical lymph nodes of patients with MS 114 as well as in mice with the model disease experimental autoimmune encephalomyelitis (EAE), 115 supporting this concept. This immunological intersection between the CNS and the peripheral immune system could thus be of great importance for initial development of this ab response in the periphery, but also for its functional properties. In this regard, we recently observed that in mice containing a high frequency of MOG-reactive T cells, the peripheral application of MOG-reactive ab triggered fulminant EAE without any further stimulation or immunization. 116 Importantly, in the absence of ongoing CNS inflammation, these molecularly large ab were unable to enter the healthy CNS; in contrast peripherally injected MOG ab triggered expansion and proinflammatory differentiation of peripheral T cells, which then infiltrated the CNS causing EAE. In an effort to understand the sequence of events leading to this unexpected finding, we observed that anti-MOG ab enabled myeloid APCs to recognize and ingest otherwise undetected traces of MOG protein via their Fc receptor. By subsequently presenting the processed antigen to T cells, these APCs then caused a fulminant activation of MOG-reactive T cells ultimately differentiating in an encephalitogenic manner. Taken together, this mechanistic observation suggests that opsonization of drained traces of CNS antigen by peripheral CNS reactive ab may be instrumental in generating and amplifying the adaptive immune response in autoimmune disorders of the CNS. In specific regard to CNS demyelinating disease associated with MOG ab, these findings may further imply that, counterintuitively, the peripheral humoral response against MOG could play a central, disease-driving role triggering acute flares of this novel disease entity.
Conclusion
MS has been and still is a heterogeneous disease. Over the last years the anti-inflammatory armamentarium for treatment of MS substantially expanded, ranging from moderately active therapeutics with few side effects to extremely efficient medications with a more severe safety profile. 117 Notwithstanding these giant advances in drug discovery, the next wave of improvement will doubtlessly generate from deciphering and delineating disease subtypes or entities within the pool of MS diagnoses, which will allow us to more precisely therapeutically counteract the respective disease-driving mechanism in an individual patient. 118 The inaugurate step in this direction was the perception and belief that NMO is not a severe variant of MS, but a disease entity in its own right. This progressive view was ultimately rewarded by the pivotal discovery of the ab response against AQP-4, which subsequently allowed us to understand NMO as an astrocytopathy, and that in this disorder peripheral B cells and ab are the prime disease drivers and accordingly the core therapeutic target.
We believe we are at the doorstep of defining the next disease entity of inflammatory CNS demyelination. Due to clinical and radiological features, most adult MOG ab+ cases were initially assigned to the loosely defined category of AQP-4– NMOSD. Re-evaluating these cases after the detection of MOG ab as the common denominator, this internally homogenous group of patients display additional characteristics, which are evidently different from AQP-4 ab+ NMO. The predominant clinical presentation of this patient group appears to be optic neuritis, in a substantial number of cases simultaneously occurring at both optic nerves; in contrast, myelitis, the cardinal symptom of NMO, is less common. Short spinal cord as well as supratentorial lesions are frequently observed on MRI scans. Most importantly, patients with MOG ab associated CNS demyelination distinctively differ from those with NMO, but even more so from those with MS, in regard to laboratory findings. CSF oligoclonal bands (OCB), the hallmark diagnostic finding in MS, appears to be rare; the more MS-specific finding within the CSF, an ab response against measles, rubella, varicella zoster virus (MRZ), is even absent. Distinctively delineating this novel patient group from NMO, AQP-4 ab are extremely rare and a coexisting autoimmune disease is uncommon in MOG ab associated disorders. The striking argument for this novel disease entity to be different from NMO derives from histopathological findings. While patients who have AQP-4+ NMO show a predominant destruction of astrocytes, all cases of MOG ab associated CNS demyelination evaluated so far revealed a vastly unaffected astrocytic structure and instead a primary demyelination of oligodendrocytes in conjunction with ab deposition and complement activation.
Specifically, this histologic finding is in line with a possible CNS demyelinating role of MOG ab in this disease. Besides this enhancing property within the CNS, anti-MOG ab, which are predominantly produced in the periphery, may exert a complementary crucial function in triggering new waves of CNS infiltration; in this regard, we have recently demonstrated that in EAE, anti-MOG ab opsonize traces of otherwise undetected MOG in secondary lymphoid organs and trigger and amplify an encephalitogenic immune response in the periphery. In conjunction with the predominant occurrence of MOG ab outside the CNS, these findings imply that, similar to NMO, MOG ab associated CNS demyelination may be primarily driven by peripheral B cells and ab; this is in turn reflected by the empiric success of plasmapheresis as a mode of acute intervention and anti-CD20-mediated depletion of B cells 119 as a prophylactic approach. If this conceptional scenario is consolidated over the next few years, innovative new therapeutic approaches, which target pathogenic B-cell function as well as ab-mediated opsonization of autoantigen, such as molecular inhibition of the Bruton’s tyrosine kinase, may be particularly fruitful.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors declare that there is no conflict of interest.