
Abstract
Introduction and purpose
Mitoxantrone (MX) is approved for the treatment of aggressive multiple sclerosis (MS) with respect to its immunosuppressive effects, especially for secondary progressive MS where treatment alternatives are sparse. Owing to its risk profile, MX use is restricted. In particular, therapy-related acute leukaemia (TRAL) has recently been discussed: whereas initial meta-analyses indicated a low risk of MX-associated TRAL in MS (0.07%) [Ghalie et al. 2002], reports of regional higher TRAL incidence have attracted considerable attention (as reviewed by Marriott et al. [2010] and Pascual et al. [2009]). A recent meta-analysis stated a risk of 0.81% of MX-associated TRAL, however, with large variability between studies [Marriott et al. 2010]. More recently prospective data from a French population (n = 802) indicated a more favourable haematologic safety profile with a TRAL incidence of 0.25% [Le Page et al. 2011]. Here we set out to evaluate the incidence of MX-associated TRAL in a large MX-treated MS cohort in a country with high MX usage.
Patients and methods
In a retrospective meta-analysis, data from six German academic MS centres were analysed (Table 1A). The observation period began between 1993 and 2005 and ended between 2007 and 2010 (median 12.0 years, 95% confidence interval [CI] 7.9–16.4). Continuous variables were expressed as means and dichotomous variables as absolute numbers and percentages. For each variable, the 95% CI was estimated. To summarize the individual results, a meta-analysis with a random effects model was computed. I2 and the p-value from the Q statistic were computed as measures for heterogeneity. All statistical analyses were performed using SAS Version 9.2 and R Version 2.10.0 (Package meta). Moreover, officially reported TRAL cases in the registry of the Drug Commission of the German Medical Association (DCGMA) and data from the German MS registry [Flachenecker et al. 2008] (and P. Flachenecker, unpublished data) are described.
Cohort characteristics and meta-analysis (A) and TRAL cases (B).
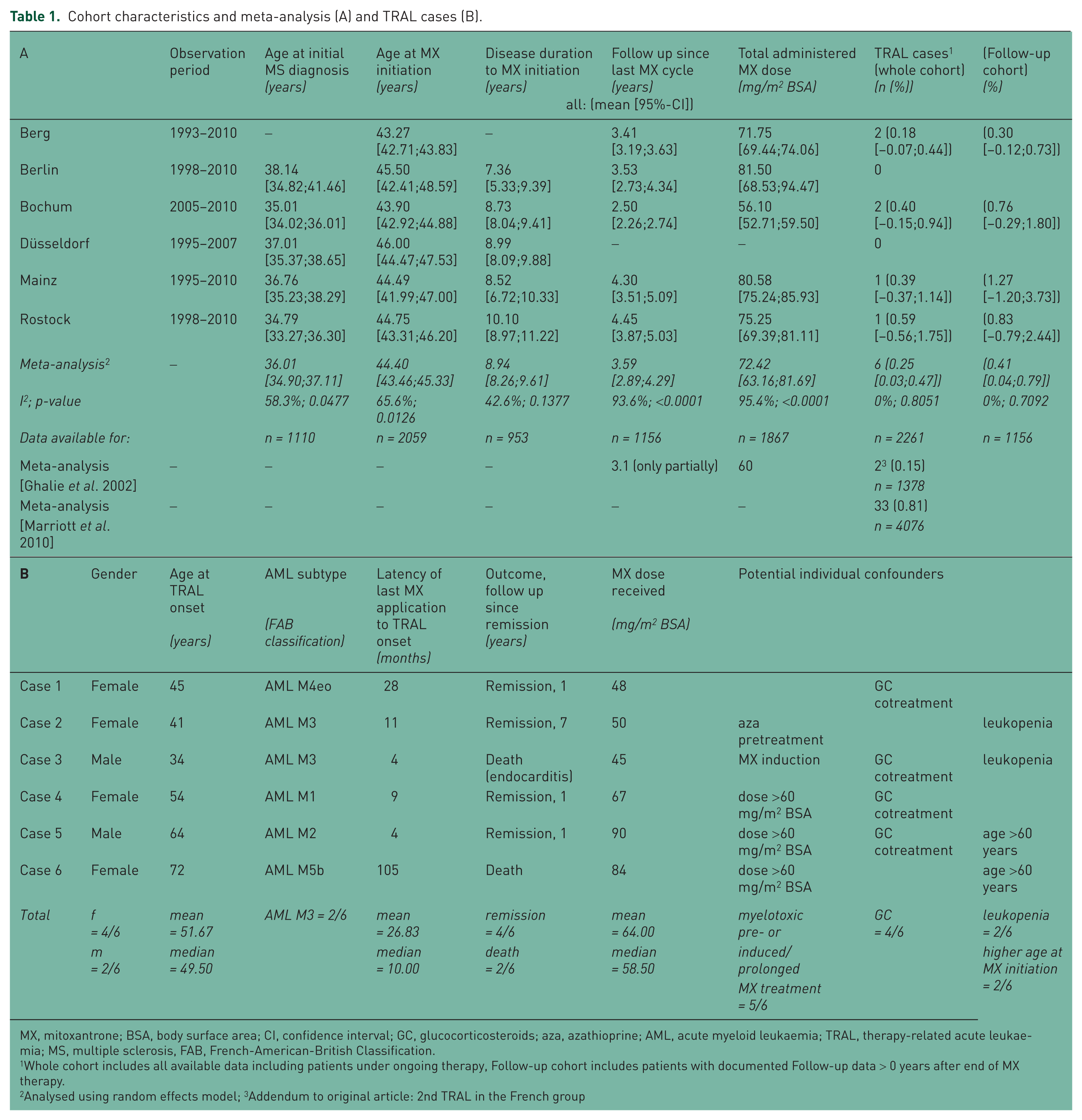
MX, mitoxantrone; BSA, body surface area; CI, confidence interval; GC, glucocorticosteroids; aza, azathioprine; AML, acute myeloid leukaemia; TRAL, therapy-related acute leukaemia; MS, multiple sclerosis, FAB, French-American-British Classification.
Whole cohort includes all available data including patients under ongoing therapy, Follow-up cohort includes patients with documented Follow-up data > 0 years after end of MX therapy.
Analysed using random effects model; 3Addendum to original article: 2nd TRAL in the French group
Results
Six TRAL cases were observed in the six major academic MS centres (Table 1A and B). When only patients are analysed who were followed up for an average of 3.6 years, an incidence of 0.41% is estimated (n = 1.156). With respect to the whole cohort of n = 2.261 patients which also includes patients under ongoing or re-initiated MX treatment (mean dose of 72.42 mg/m2 body surface area [BSA]), an incidence of 0.25% is calculated.
TRAL onset was on average 26.8 months following the end of MX treatment. Two promyelocytic acute myeloid leukaemias (AML M3) and one case each of AML M1, M2, M4eo and M5b were diagnosed. Two AML ended fatal, four went into remission. Cumulative dosage in TRAL patients varied between 45 and 90 mg/m2 BSA (mean 64 mg/m2, median 58.5 mg/m2). Despite dose adaption according to leukocyte counts, two cases exhibited pronounced leukocyte nadirs with normalization before next MX cycle before AML onset (Table 1B). In case 3, leukopenia did not resolve within 3 months after the fifth MX cycle. Case 2 had received azathioprine (100 mg/day) for 3 years, case 3 had received MX induction therapy for the first two cycles (8-week interval, cumulative 22 mg/m2 BSA). Four of six TRAL patients had been cotreated with glucocorticosteroids (GCs; intravenous or intrathecal) with each MX cycle on a regular basis.
Between 1990 and August 2010, 11 TRAL cases were reported to DCGMA. With data from the German MS registry that on average 8.4% (2006: 10.4%, 2007: 8.8%, 2008: 6.9%, 2009: 7.5%) of all MS patients were treated with MX and approximately 122,000 MS patients in Germany [Flachenecker et al. 2008] (and P. Flachenecker, unpublished data), estimated frequency of TRAL ranges between 0.09% and 0.13%.
Discussion and conclusions
Using two different approaches, we observe a considerably lower MX-associated TRAL incidence in Germany than in recent reports from other regions (as reviewed by Marriott et al. [2010] and Pascual et al. [2009]). These in majority retrospective reports with low patient numbers offered partially incomplete data, e.g. concerning dosage, potential risk factors and diagnosis of leukaemia. Importantly, treatment protocols differ concerning intervals, dosages and cotreatments and were mostly without titration according to leukocyte counts as performed in the pivotal phase III trial [Hartung et al. 2002]. In a prospective albeit small study (n = 230, TRAL incidence 2.82%), MX therapy in five of six TRAL cases was initially administered in monthly intervals (10 mg/m2 BSA) independent from leukocyte nadirs, and two TRAL patients had received azathioprine before or after MX treatment [Pascual et al. 2009]. As two TRAL patients in our series had either received MX induction therapy or azathioprine treatment, higher dosage over shorter time and myelotoxic cotreatments may represent independent risk factors. Concomitant GC treatment may pose an additional confounder since four cases had received GC with each MX cycle and GC have been demonstrated to increase intracellular MX concentration in vitro [Cotte et al. 2009]. However, in a prospective French study [Le Page et al. 2011] where the majority of the patients were treated with a monthly combination of MX and GC for a mean cumulative mitoxantrone dose of 78 mg/m2 BSA a comparable TRAL incidence (0.25%) was observed (mean follow-up duration: 6.7 years).
Data on AML subtypes, outcome and latency to TRAL onset in our cohort (Table 1B) is comparable with previous reports [Pascual et al. 2009; Ellis and Boggild, 2009] and compatible with haematological data on secondary leukaemia after topoisomerase-II-inhibitor treatment (latency 0.5–5 years). At TRAL onset, three of six patients exhibited a lower cumulative dosage than previously suggested as a risk factor (60 mg/m2 BSA) [Ellis and Boggild, 2009]; thus, a hypothetical threshold dosage still needs to be defined. In two cases, dose adjustment due to pronounced leukocyte nadirs did not prevent TRAL development. These aspects might point to individual predisposition and argue for rapid bone marrow biopsy in long-lasting cytopenia despite dose reduction. Currently, potential pharmacogenetic risk factors [Cotte et al. 2009] are being investigated in a prospective study.
Despite the large cohort observed, our data has to be interpreted with caution due to the retrospective approach with different observation periods. Still, more recently two prospective albeit smaller studies have demonstrated TRAL incidences similar to the range reported here [Le Page et al. 2011; Rivera et al. 2009].
In summary, large regional variability of TRAL incidence may point to other confounding factors such as administration protocols and cotreatments. This emphasizes the urgent need for uniform treatment algorithms in addition to thorough postmarketing surveillance [Marriott et al. 2010] including ongoing haematologic screening for at least 5 years after the end of MX treatment [Le Page et al. 2011].
Footnotes
This work was supported by grants from the German Ministry for Education and Research (BMBF, ‘German Competence Network Multiple Sclerosis’ [KKNMS], CONTROL MS, 01GI0914 to RG and AC) and from the German Research Foundation (DFG Exc 257 to JD and FP).
A. Stroet, C. Hemmelmann, M. Starck, P. Flachenecker, V. Fleischer, H. Nückel and A. Ziegler have nothing to disclose. U. Zettl has received research support from Bayer Healthcare, Biogen Idec, Merck Serono and Teva Neuroscience. J. Dörr has received research support and personal compensation for activities with Novartis and Bayer Schering. F. Paul has received personal compensation for activities with Bayer Schering, Merck Serono, Teva Neuroscience and Novartis and research support from Bayer Schering, Merck Serono, Teva Neuroscience and Novartis. F. Paul has received compensation and/or their research work has been funded, entirely or in part, by a grant to their university. The grant agreement requires that the name of the funding entity and the purpose of the grant may not be disclosed. The funding entity is a governmental organization. F. Zipp has received research grants from Johnson & Johnson, Teva, Merck Serono, and Bayer as well as consultation funds from Johnson & Johnson. R. Gold has received personal compensation for activities with Bayer Healthcare, Biogen Idec and Teva Neuroscience and in an editorial capacity from Therapeutic Advances in Neurological Disorders, and also received patent payments from Biogen Idec and research support from Bayer Healthcare, Biogen Idec, Merck Serono, Teva Neuroscience and Novartis. B. Kieseier has received personal compensation for activities with Bayer Schering, Biogen Idec, Merck Serono, Novartis, Roche, Sanofi Aventis and Teva Neuroscience and research support from Bayer Schering, Biogen Idec, Merck Serono and Teva Neuroscience.
A. Chan has received personal compensation for activities with Bayer Schering, Biogen Idec, Merck Serono and Teva Neuroscience and research support from Bayer Schering, Biogen Idec, Merck Serono and Novartis.