
Abstract
Background:
PF-06687234 is a novel, human, single-chain variable fragment cytokine fusion protein comprising the antibody fragment F8 and the immunoregulatory cytokine interleukin-10.
Objectives:
We evaluated the efficacy and safety of PF-06687234 as an add-on therapy to infliximab in participants with active ulcerative colitis (UC).
Design:
This phase IIa, double-blind, placebo-controlled, parallel-group, multicenter study randomized (1:1) participants with active UC to subcutaneous PF-06687234 20 mg or placebo once weekly, and participants continued intravenous background infliximab every 6 or 8 weeks, for 12 weeks.
Methods:
The primary endpoint was the proportion of participants who achieved clinical remission determined by the modified total Mayo score at week 12. Safety assessments, including adverse event (AE) reporting and laboratory abnormalities, were also primary endpoints. Secondary endpoints included the proportion of participants achieving endoscopic improvement at week 12, and exploratory endpoints included the concentration of PF-06687234 in colonic mucosal tissue from biopsies.
Results:
After 20 participants were randomized and treated, this study was stopped early for futility. There were no statistically significant differences between groups for any efficacy endpoint (all p > 0.05), although numerical trends towards efficacy were observed in some secondary endpoints. PF-06687234 was detected at a low concentration in only one colon tissue sample. There were no differences in AEs between the groups. The most frequently reported AE in the PF-06687234-treated group was injection site reaction.
Conclusion:
PF-06687234 was well tolerated, but when combined with background infliximab, did not meet the primary efficacy endpoint in this cohort of participants with active UC. Efficacious PF-06687234 tissue concentrations may not have been achieved at the dose tested.
Trail registration:
Efficacy, safety, and tolerability of PF-06687234 as add-on therapy to infliximab in active UC subjects not in remission. https://clinicaltrials.gov/study/NCT03269695. NCT03269695.
Introduction
Ulcerative colitis (UC) is a chronic inflammatory bowel disease (IBD) that affects the intestinal mucosa surface, starting in the rectum and progressing to the more proximal parts of the colon. 1 Anti-tumor necrosis factor (TNF)-α antibodies, such as infliximab, adalimumab, and golimumab, and the α4β7 integrin antibody vedolizumab, are often used as first-line biological treatments in patients with no response or side effects to primary treatments.2–6 Other current treatment options include the monoclonal antibody against the p40 subunit of interleukin (IL)-12 and IL-23 ustekinumab, 7 the small molecule Janus kinase (JAK) inhibitors tofacitinib, filgotinib, and upadacitinib,8–10 and the sphingosine 1-phosphate receptor modulator ozanimod. 11
Despite advances in understanding the pathophysiology of UC and the introduction of new therapeutic options, long-term remission rates remain low, at approximately 20% to 30%, suggesting the possible existence of a “therapeutic efficacy ceiling” for current treatments. 12 Furthermore, some patients do not respond to initial treatment (primary nonresponders), while others lose their response over time (secondary nonresponders). 13 Not all primary responders reach the target of remission, 14 and nonremission rates to anti-TNF-α agents in patients with IBD range from 63% to 78% at week 52–54.2,4,15
IL-10 is a master regulatory cytokine that exhibits a wide range of pro-inflammatory and anti-inflammatory effects on the innate and adaptive immune system, 16 including upregulation of interferon (IFN)-γ and neopterin at high doses. 17 IL-10 has been widely studied in the context of IBD.18,19 In this study, it was hypothesized that increasing the levels of IL-10 in the tissues of patients with active UC would lead to the restoration of immunoregulatory balance, thereby reducing tissue inflammation. One potential innovative treatment approach to test this “IL-10 hypothesis” that could improve the response rate, break the “efficacy ceiling,” and enhance the persistence of response compared with existing therapeutic options, is a combination therapy that could harness synergistic mechanisms of action, possibly leading to a larger and more sustained clinical response over time. One approach to such combination therapy is PF-06687234 (also known as Dekavil or IL10-F8), a novel, human, single-chain variable fragment fusion protein comprising the antibody fragment F8 and IL-10. F8 selectively binds to fibronectin extra-domain A, which is primarily expressed in blood vessels during inflammation. 20 The inclusion of the F8 component in PF-06687234 is designed to enable the retention of IL-10 at sites of active inflammation in the tissues of patients with active UC. Previous trials have assessed recombinant human IL-10 as a treatment for Crohn’s disease (CD), another type of IBD. Subcutaneous (SC) administration of once-daily IL-10 in patients with mild-to-moderate active CD showed clinical and endoscopic improvement, while in patients with chronic active CD, a tendency toward clinical improvement but not remission was observed.21,22 Several factors may have contributed to the inconclusive results regarding the clinical benefit of IL-10 in these studies, including colonic tissue concentrations of IL-10 being insufficient to elicit a response, the short plasma half-life of recombinant IL-10, the dose-limiting tolerability (anemia and thrombocytopenia) of IL-10 administration, and the resistance of the lamina propria mononuclear cells to IL-10.18,22,23 These limitations emphasize the need for alternative strategies that can enrich IL-10 concentration in inflamed intestinal tissues, which could potentially lead to greater clinical efficacy.
The conjugation of IL-10 and F8 is intended to increase the retention of the recombinant cytokine at sites of inflammation and could offer a promising solution to overcome these challenges. Notably, IL10-F8 has been shown to be retained in arthritic lesions of mice with collagen-induced arthritis, effectively limiting disease progression. 24 This selective retention has also been observed in patients with rheumatoid arthritis, 25 with emerging clinical data supporting its therapeutic potential. 26 Furthermore, in contrast to anti-TNF-α biologic agents, which rely on direct inhibition of pro-inflammatory cytokines, PF-06687234 is hypothesized to exert clinical benefits through its direct agonist effect on IL-10 receptors and subsequent modulation of the innate and adaptive immune system to restore immune homeostasis. Indeed, a synergistic effect has been demonstrated in a preclinical animal model of arthritis in which an anti-TNF-α agent and IL10-F8 were administered concomitantly. 27 Therefore, combining PF-06687234 with existing anti-TNF-α therapies may offer additive or synergistic clinical benefits, which is particularly relevant considering the high rates of primary nonresponse and nonremission to anti-TNF-α agents in patients with IBD. 14 By utilizing PF-06687234 in combination with infliximab, there is potential to restore patients’ response to infliximab treatment.
Taken together, these findings suggest that PF-06687234 could be a potential treatment for UC, either as a standalone therapeutic or as part of a combination regimen that can overcome the dose-limiting toxicity of IL-10. In this study, the aim was to assess the efficacy and safety of PF-06687234 as an add-on therapy in patients with active UC and a nonremission (partial) response to infliximab.
Materials and methods
Study design
Study B7581002 (NCT03269695) was a phase IIa, randomized, double-blind, placebo-controlled, parallel-group, multicenter study conducted at 27 sites across 11 countries in participants with active UC and prior partial response but nonremission to infliximab. The study comprised a 4-week screening period, a 12-week treatment period, and a 10-week follow-up period (Figure 1). Eligible participants were randomized (1:1) to SC PF-06687234 20 mg and intravenous (IV) infliximab (referred to as the “PF-06687234 plus infliximab” group) or placebo and infliximab (referred to as the “infliximab-only” group). PF-06687234 or placebo was administered as an SC injection once weekly. Infliximab was administered every 6 or 8 weeks to allow participants to continue their stable infliximab regimen.
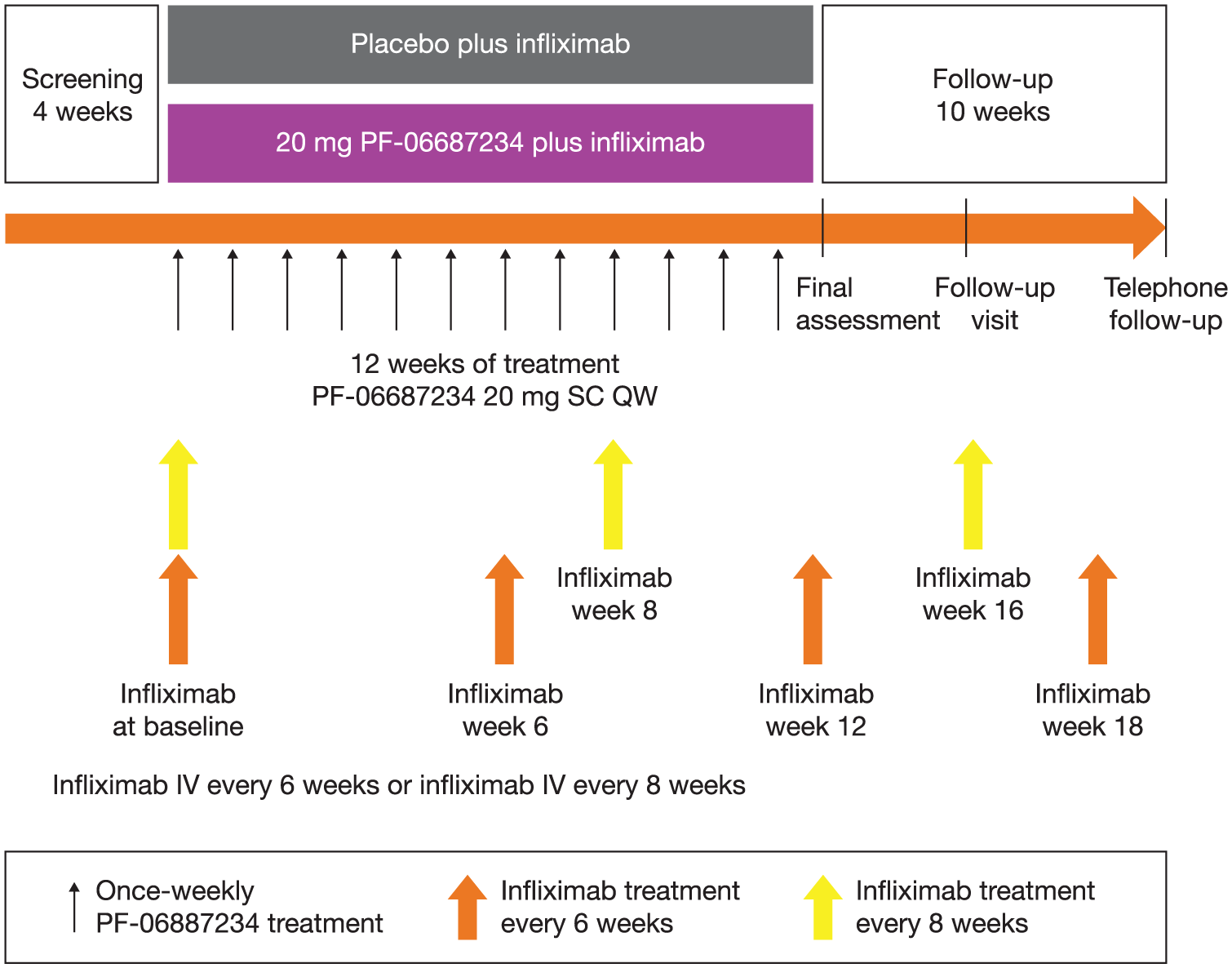
Study design.
The dose of PF-06687234 (20 mg SC weekly) was selected since it was expected to result in free IL-10 concentrations comparable to those achieved with recombinant human IL-10 and associated with signals of efficacy in patients with Crohn’s disease.21,22
The reporting of this study conforms to the Consolidated Standards for Reporting Trials (CONSORT) statement. 28
Patients
Male and female participants aged 18–75 years, with a body weight >40 kg, a diagnosis of active UC for ⩾4 months, defined by a total Mayo score ⩾4 but ⩽9 and an endoscopic subscore ⩾2, UC extending at least 15 cm proximal to the anal verge at the time of screening endoscopy, a partial response to infliximab (or infliximab biosimilar), and who were receiving a stable dose (5–10 mg/kg) of infliximab (or infliximab biosimilar) for a minimum of 14 weeks prior to the start of the study, were included. Determination of a partial response to infliximab was left to the discretion of the investigator at each site. Guidance was provided in the study protocol that, for a participant to be considered eligible for inclusion, they must have received infliximab (or biosimilar) for at least 14 weeks, have experienced some degree of clinical improvement, but still meet the eligibility criteria for disease severity at the screening and baseline visits. Patients receiving oral 5-aminosalicylic acid or sulfasalazine for at least 4 weeks prior to baseline, oral corticosteroids (prednisone equivalent up to 20 mg/day or budesonide up to 9 mg/day) for at least 2 weeks prior to baseline, or 6-mercaptopurine, azathioprine, or methotrexate for at least 8 weeks prior to baseline, as long as doses remained stable throughout the study, were also eligible.
Exclusion criteria included diagnosis or documented history of total colectomy and/or pouchitis, indeterminate colitis, microscopic colitis, ischemic colitis, infectious colitis, radiation colitis, and diverticular disease associated with colitis, or clinical findings suggestive of CD; a history of, or at screening, endoscopy, biopsy-documented colonic dysplasia or neoplasia; requirement of infliximab (or infliximab biosimilar) dosing interval other than every 6 or 8 weeks during the study; clinical signs of fulminant colitis or toxic megacolon; primary sclerosing cholangitis; known colonic stricture, or history of colonic or small bowel obstruction or resection; history of, or current, colonic or small bowel stoma. Additional exclusion criteria included any cardiovascular, renal, hepatic, hematologic, gastrointestinal, endocrine, pulmonary, immunologic, or neurologic condition; any clinically significant infection within 6 months of baseline; history of cancer within the previous 5 years; use of oral systemic corticosteroids (>9 mg/day of oral budesonide or >20 mg/day of prednisone or equivalent), anti-integrin inhibitors, IFN therapy, lymphocyte-depleting agents, or JAK inhibitors.
Endpoints
Primary endpoints
The primary endpoint was the proportion of participants who achieved clinical remission at week 12, as confirmed by the modified total Mayo score, which consists of three domains: endoscopy, stool frequency, and rectal bleeding. Remission was defined as a traditional endoscopic subscore ⩽1 (where mild friability was scored as 1 and moderate or severe friability was scored as 2), stool frequency subscore ⩽1, and rectal bleeding subscore = 0. 29 Safety assessments were also considered a primary endpoint and included the incidence and severity of adverse events (AEs), serious AEs (SAEs), and withdrawals due to AEs; laboratory abnormalities, changes in vital signs, and electrocardiogram (ECG) findings.
Secondary and exploratory endpoints
Secondary endpoints included the proportion of participants with endoscopic improvement at week 12, defined as a decrease of ⩾1 point in modified Mayo endoscopic subscore (which scores any friability as 2) or an absolute Mayo endoscopy subscore of ⩽1 without friability. Other secondary endpoints were the proportion of participants achieving histologic remission defined by Geboes score (Grade 3 ⩽3.1) and Robarts histology indices (RHI; ⩽5) at week 12. 30 Geboes Grade 3 ⩽3.1 score was defined as <5% crypts involving neutrophilic infiltration in the epithelium. 31
Secondary endpoints also included the change from baseline (CFB) in fecal calprotectin at week 11 and CFB in high sensitivity C-reactive protein (hsCRP) at weeks 4, 8, and 11, the proportion of participants with a clinical response at week 12 (defined as a decrease from baseline of ⩾3 points in total Mayo score with ⩾30% change, accompanied by ⩾1-point decrease or absolute score of 0 or 1 in rectal bleeding subscore), 29 the proportion of participants with a CFB in partial Mayo score ⩽2 with no individual subscore >1 at weeks 2, 4, 6, 8, and 12, and the proportion of participants achieving mucosal healing at week 12 (defined as endoscopy subscore ⩽ 1).
Pharmacokinetics (PK) outcomes were exploratory and included the concentration of PF-06687234 in serum and in biopsies of inflamed and noninflamed colonic tissue. Colonic tissue biopsies were collected during endoscopic procedures at screening and at week 12. A total of 14 biopsies were taken at both time points for each participant: 11 from inflamed colonic mucosa and three from normal-appearing colonic mucosa in a targeted manner from the most affected area 15–30 cm from the anal verge in the colon. Four samples were analyzed from each of 16 participants, which included inflamed and noninflamed tissue at screening and at week 12. One participant had three replicates of inflamed tissue at week 12 for a total of 66 samples analyzed. Several of the biopsies were combined to obtain sufficient material to enable the assay. Concentrations of PF-06687234 in tissue were measured at week 12 with a specifically developed and validated novel fully quantitative peptide immuno-affinity liquid chromatography–tandem mass spectrometry (LC-MS) method. This method is described briefly as follows: following tissue homogenization, protein extraction, and trypsin digestion, the proteotypic peptide, GRPPTFGQGTK, which uniquely represents PF-06687234, was extracted using an anti-peptide antibody prior to LC-MS quantification. This approach not only ensures high measurement selectivity and sensitivity but also overcomes bioanalytical challenges associated with other assay formats that rely on antibody capture from detergent-containing tissue lysate. Additional LC-MS methodology details are provided in the Supplemental Material.
Serum for analysis of PF-06687234 concentration was collected approximately 30 min prior to dosing and at weeks 1, 3, 7, 11, and 12. A subgroup of participants consented for additional PK sampling at day 1 and week 11 to allow for the calculation of: area under the concentration–time profile from time 0 to time tau (tau), dosing interval, where tau = 168 h (AUCtau), maximum serum concentration (Cmax), and time for Cmax (Tmax).
Additional secondary endpoints included the incidence of the development of human anti-fusion antibodies (HAFAs) and neutralizing antibodies (NAbs) against PF-06687234, evaluated at day 1 and weeks 3, 7, 11, and 12.
Statistical analyses
The sample size was based on the primary efficacy endpoint. It was anticipated that 22% of participants in the infliximab-only group and 52% of participants in the PF-06687234 plus infliximab group would achieve clinical remission at week 12. With a 1:1 randomization ratio, a sample size of 64 participants (32 participants in each treatment group) was required to detect a treatment advantage of 30 percentage points in clinical remission for the active treatment combination, with at least 80% power and a one-sided type-I error at 0.05. Assuming a 15% dropout rate, the total target sample size for recruitment was 76 participants.
A protocol-specified interim analysis (IA) for futility was planned at 40% enrollment; however, due to extremely slow recruitment over more than 2 and half years, resulting in n = 17, a new IA for futility was established. The initial IA criteria, based on a single primary endpoint, would have been highly unreliable in such a small sample size, since one or two outcomes could disproportionally influence the probability of success. Consequently, a new IA futility approach was implemented using four criteria. These were based on four endpoints (reflecting the totality of the data), with control over two error types: a false IA stopping rate (1-specificity) <20% and a false negative rate (1-sensitivity) <50%. It was determined that further enrollment could continue if the following probability of success (conditional power) criteria were met for all four endpoints, simultaneously: >32% modified remission, >30% remission, and >36% mucosal healing at week 12, and >10% decrease in fecal calprotectin at week 11. Controlling both error types in decision-making enhances the robustness of the IA futility decision rule. For analyses with binary data, participants with missing values for any reason were considered as treatment failures.
Binary data were analyzed using the unconditional exact Chan and Zhang method. 32 Longitudinal continuous data were analyzed with mixed effect model repeated measurement, and nonlongitudinal continuous data were analyzed by analysis of covariance.
Efficacy, pharmacodynamics, and biomarker analyses were performed in the modified intention-to-treat population, defined as all participants who were randomized and who received at least one dose of the study treatment. The safety population included all participants who received at least one dose of the study treatment, and safety data were summarized descriptively. The PK population was defined as all participants who received at least one dose of PF-06687234 and had data on at least one PK concentration, and the immunogenicity assessment population was defined as all enrolled participants who received at least one dose of PF-06687234 with at least one post-treatment HAFA determination.
Results
Patients
In total, 20 participants (10 participants in each treatment group) were enrolled and treated (Figure 2) prior to the scheduled IA. Of these participants, 15 (75.0%) participants completed treatment; 7 (70.0%) in the PF-06687234 plus infliximab group and 8 (80.0%) in the infliximab-only group. A total of five (25.0%) participants (PF-06687234 plus infliximab group (n = 3) and infliximab-only group (n = 2)) discontinued treatment, mostly due to AEs (n = 3; 15.0%), but continued to participate in the study.

Patient disposition.
Demographic and baseline disease characteristics were generally balanced between the two treatment groups (Table 1). The mean age of participants was 44.6 years. All participants were diagnosed with UC, with a median (range) duration of disease of 7 (1–16) years. Mean body mass index was slightly higher in the PF-06687234 plus infliximab group. One participant in the infliximab-only group did not experience a response to prior anti-TNF therapy. Prior corticosteroid use was higher in the PF-06687234 plus infliximab group than in the infliximab-only group. There were more participants with fecal calprotectin levels >250 μg/g in the PF-06687234 plus infliximab group than in the infliximab-only group. The median baseline fecal calprotectin for the infliximab-only group was 359.5 μg/g and for the PF-06687234 plus infliximab group was 907.0 μg/g. The extent of disease, total Mayo score, and partial Mayo score at baseline were comparable between groups.
Patient demographics and baseline disease characteristics (safety population).

The safety population included all participants who received at least one dose of the study treatment.
Included for continuous variables only.
Based on titer ⩾1.30.
5-ASA, 5-aminosalicylic acid; ADA, anti-drug antibodies; BMI, body mass index; N, the number of patients in the safety analysis set within each treatment group; n, the number of patients in each demographic or with each characteristic; SD, standard deviation; TNF, tumor necrosis factor.
At baseline, five participants (25%) were receiving infliximab at doses >5 mg and/or with dosing intervals shorter than 8 weeks, indicating optimized infliximab treatment prior to study entry.
Efficacy
Primary endpoints
Modified clinical remission was achieved by 14.3% and 12.5% of participants in the PF-06687234 plus infliximab and infliximab-only groups, respectively, at week 12. No significant difference was observed between the two groups (Figure 3(a); one-sided p value: 0.5486).

Proportion of participants with (a) modified clinical remission and (b) endoscopic improvement (mITT population).
Secondary and exploratory endpoints
Numerically more participants achieved endoscopic improvement (decrease ⩾1 point in modified endoscopic subscore or an absolute endoscopy score ⩽1) at week 12 in the PF-06687234 plus infliximab group, compared with the infliximab-only group (57.1% vs 25.0%; one-sided p value: 0.1583; Figure 3(b)); however, no significant difference was observed between the two groups.
The proportion of participants achieving Geboes histology index remission at week 12 was not statistically different between groups (PF-06687234 plus infliximab group: 37.5%; infliximab-only group: 62.5%; one-sided p value: 0.7393). Similarly, the proportion of participants achieving RHI histology remission at week 12 was not significant between groups (PF-06687234 plus infliximab group: 37.5%; infliximab-only group: 50.0%; one-sided p value: 0.5982).
There was no significant difference in mean CFB in fecal calprotectin levels at week 11 between the PF-06687234 plus infliximab (−109.02 µg/g) and infliximab-only (−378.63 µg/g) groups (two-sided p value: 0.3525). There were no statistical or clinically meaningful CFB differences in hsCRP levels between the PF-06687234 plus infliximab and infliximab-only groups at weeks 4, 8, and 11.
A numerically greater proportion of participants achieved a clinical response (⩾3-point decrease from baseline in total Mayo score with ⩾30% change, accompanied by ⩾1-point decrease from baseline in rectal bleeding subscore or an absolute rectal bleeding subscore of 0 or 1) at week 12 in the PF-06687234 plus infliximab group (85.7%) compared with the infliximab-only group (37.5%). However, a statistical difference between the two groups was not observed (two-sided p value: 0.0732). No statistically significant differences were observed between the PF-06687234 plus infliximab and infliximab-only groups in the proportion of participants with a CFB in partial Mayo score ⩽2 with no individual subscore >1 at weeks 2, 4, 8, and 12 (Table 2).
Proportion of participants with a change from baseline in partial Mayo score ⩽2 with no individual subscore >1 up to week 12 (mITT population).

The mITT population included all participants who were randomized and received at least one dose of the study treatment. Generalized linear mixed model was used with fixed effects of treatment, visit, and treatment-by-visit interaction. The RMPL estimation method was used. Baseline was defined as the last measurement prior to the first dosing (day 1).
CI, confidence interval; mITT, modified intention-to-treat; RMPL, residual pseudo-likelihood.
Only one participant in the PF-06687234 plus infliximab group achieved mucosal healing (defined as endoscopy subscore ⩽ 1, where any friability was scored as 2) at week 12, while none did in the infliximab-only group; this difference was not significant (two-sided p value: 0.4700).
However, modified clinical remission, clinical remission, mucosal healing, and fecal calprotectin levels were not able to meet the pre-specified endpoint requirements at the IA, and therefore, the study was terminated for futility.
Pharmacokinetics
Three participants treated with PF-06687234 provided consent to participate in the PK subgroup, which involved additional sampling. All samples collected from these participants were analyzed, and PK parameters were calculated. One of these participants had a concentration below the limit of detection (0.313 ng/mL) and was therefore not included in the PK analysis. Sparse PK samples collected from other participants during the course of the study were also summarized separately.
Serum pharmacokinetics
At day 1, following a single dose of PF-06687234, individual Cmax was observed at 21.3 and 21.7 hours post-dose in the PK subgroup. In those two participants, Cmax was observed at 22.9 and 23.8 hours post-dose at week 11. Exposure based on individual AUCtau and Cmax values varied between the two participants in the PK subgroup, with the greatest differences observed at week 11, with a 44- and 101-fold difference, respectively.
The individual pre-dose serum PF-06687234 concentration–time profiles for PF-06687234 at day 1, week 1, week 3, week 7, and week 11 in participants with sparse PK samples are shown in Supplemental Figure S1. Serum concentrations of PF-06687234 were low.
Colonic mucosa pharmacokinetics
Four samples were analyzed from each of 16 participants, with one participant having three replicates of inflamed tissue at week 12 for a total of 66 samples analyzed. Out of these 66 samples, the concentration of PF-06687234 in biopsies from colonic mucosa was above the limit of quantification (1.56 ng/mL tissue lysate) in only one sample. The sample was from a biopsy of noninflamed tissue, and a concentration of PF-06687234 of 2.85 ng/mL of lysate was measured while the protein concentration was 5.099 mg/mL.
Immunogenicity
Following the administration of PF-06687234, a few participants developed anti-PF-06687234 antibodies, although none of them tested positive for anti-PF-06687234 IL-10 NAbs. Two participants tested positive for anti-PF-06687234 F8 single-chain variable fragment NAbs at week 7, although the positive response was no longer detectable in either of the participants at week 11 and at later time points.
Although a few participants developed HAFAs and NAbs against the F8 portion of PF-06687234, these data, while limited, do not seem to correlate with lower serum concentration (data not shown).
Safety
Overall, 17 (85.0%) participants reported a total of 45 treatment-emergent AEs (TEAEs), 10 of which were considered treatment-related. The incidence of TEAEs was similar between the two treatment groups, and the majority of TEAEs were mild in severity. Two (10.0%) participants, both in the infliximab-only group, had severe TEAEs. One participant had an SAE of worsening of UC, and one participant had severe nonserious AEs of worsening of UC and acne.
A total of three (15.0%) participants permanently discontinued from treatment due to AEs. One participant in the PF-06687234 plus infliximab group discontinued due to a mild TEAE of diabetic nephropathy. Two participants in the infliximab-only group discontinued treatment; one due to a worsening moderate AE of UC that was considered to be treatment-related and another due to an SAE of relapsing active UC, which in this case was considered unrelated to treatment.
The most frequently reported TEAEs were injection site reaction and UC in the PF-06687234 plus infliximab and infliximab-only groups, respectively (Table 3). Two participants in the overall study population reported moderate thrombocytopenia and anemia, both in the PF-06687234 plus infliximab group (n = 1 (5%) for each).
Incidence of TEAEs (all-causalities) by system organ class (safety population).

The safety population included all participants who received at least one dose of the study treatment.
TEAE, treatment-emergent adverse event.
A total of 18 (90.0%) participants had laboratory test abnormalities. The most common clinically significant laboratory abnormality was leukocyte esterase ⩾1, reported by a total of seven (35.0%) participants (four in the infliximab-only group and three in the PF-06687234 plus infliximab group). In addition to the laboratory findings, one urinary tract infection TEAE was clinically observed (in the infliximab-only group). There were no deaths reported, no clinically significant trends observed in vital signs data, and no clinically significant ECG abnormalities.
Discussion
The clinical impact of IL-10 on IBD is unknown, as studies in CD have yielded inconclusive results.21,22 One potential explanation for these findings is the inability to deliver sufficient concentrations of IL-10 to the colonic mucosa to produce an immunoregulatory effect at the sites of inflammation. Alternative strategies have been explored to address this challenge, including the oral administration of genetically modified bacteria to directly deliver IL-10 to the gut lumen, 33 or the coupling of IL-10 to a transporter protein (AMT-101) to facilitate IL-10 delivery to the intestinal mucosa following oral administration. 34 The bacterial vector approach has demonstrated efficacy in animal models of colitis, 35 although clinical data are awaited. Notably, clinical data are beginning to emerge for AMT-101, showing promising signs of efficacy in chronic pouchitis. 36
Our approach to deliver IL-10 to the sites of inflammation in the colonic mucosa employed PF-06687234, a protein comprising the antibody fragment F8 and the immunoregulatory cytokine IL-10. We assessed the clinical potential of PF-06687234 as an add-on therapy to infliximab in participants with UC who were not in remission. However, in this clinical study, despite numerical differences favoring PF-06687234, no statistically significant difference was observed in participants achieving remission or clinical improvements between the PF-06687234 plus infliximab and infliximab-only groups. PF-06687234 was generally well tolerated and no safety findings of note were identified, although two participants in the PF-06687234 plus infliximab group experienced moderate cases of thrombocytopenia (one participant) and anemia (one participant).
This study experienced great difficulty in recruiting participants according to the eligibility criteria of the trial and was halted due to futility following an IA because it was deemed statistically improbable to achieve the predetermined efficacy endpoints of the study, were it to have continued to completion. There are several potential reasons for these findings. Firstly, the lack of a standard definition of “partial responders” necessitated the need for a study-specific definition being employed. This approach may have contributed to heterogeneity among participants at baseline, particularly as magnitude of prior benefit to infliximab was not captured, and reasons for partial response were not fully understood. However, heterogeneity was not evident from participant baseline characteristics. Additional heterogeneity may have been introduced as a consequence of prior treatment history. In the PF-06687234 plus infliximab group, three participants had received prior treatment with golimumab, and three participants had received prior treatment with vedolizumab; in the placebo plus infliximab group, two participants had received prior vedolizumab.
Concentrations of PF-06687234 were low (and variable) in serum and also low or undetectable in the target tissue without any apparent correlation with HAFAs or NAbs. This suggests weak F8 binding at the target site (inflamed tissue) in the intestine. These observations contrast with those observed in preclinical models, where target tissue exposure has been reported to be multiple times higher than serum concentrations, 27 and also from clinical data confirming the retention of IL10-F8 at the inflamed joint in participants with rheumatoid arthritis. 25 Consequently, the localization of PF-06687234 to the inflamed colonic mucosa could not be established in participants with UC in this study. This may be due to the dosing regimen used in this study and the timing of sample collection. Lastly, although the novel data analysis method was designed to minimize false positives, the risk of false negatives remained high. Our findings differ from those emerging for AMT-101, a chimeric fusion protein designed to be gut restricted, 34 which has shown clinical efficacy and anti-inflammatory responses in patients with chronic pouchitis. 36 However, AMT-101 treatment in patients with moderate-to-severe UC, either as monotherapy or when given in combination with adalimumab, did not show clinical efficacy.37,38 These differences suggest that local delivery of IL-10 from the intestinal lumen may be an effective treatment in selected conditions and this approach needs further exploration.
In clinical practice, combination therapy is more commonly used in patients with UC or CD who have not responded adequately to previous monotherapies; however, there are currently limited clinical trials examining combination therapy. The SONIC trial, conducted in patients with CD who had not received prior biologic or immunosuppressive treatment, compared the efficacy of infliximab or azathioprine monotherapy with combination treatment and demonstrated the superiority of the combination therapy. 39 More recently, the VEGA study investigated the efficacy of SC TNF-α blocker golimumab and IV IL-23 antagonist monoclonal antibody guselkumab as both monotherapy and in combination. Although there was a numerical superiority for the combination treatment, in terms of efficacy endpoints, the differences between the combination therapy and the monotherapy groups did not reach statistical significance for guselkumab, but did reach statistical significance for golimumab. It is worth noting that the participants included in the VEGA trial had no prior exposure to the classes of drugs under investigation, which differs from the present study. 40
The present study included some important strengths. This is the first study in participants with UC that evaluated PF-06687234, a novel antibody–cytokine fusion protein with the potential to harness the therapeutic activity of the active cytokine moiety at the site of disease, while limiting safety and tolerability issues associated with systemic exposure. The design of the trial, which includes partial responders to previous treatment and placebo-controlled combination treatment with a baseline biological therapy like anti-TNF-α, could be utilized for future trials with agents in combination. The study also employed an innovative statistical analysis, handling drop-out rates with missing values through a treatment failure approach for binary endpoints and observed cases for continuous endpoints, while also conducting an IA to assess futility and facilitate decision-making with a limited amount of data. The pre-planned IA for futility allowed the study to be halted once the pre-specified decision criteria were met, indicating that primary endpoints were unlikely to be achieved and, as a result, further participants were spared from exposure to a drug that showed suboptimal efficacy in this setting. 41 We were unable to demonstrate clinically relevant concentrations of PF-06687234 in serum or colonic tissue, despite the use of a novel bioanalytical LC-MS methodology for the latter, which was contrary to our hypothesis and unexpected. Since colonic samples were only collected at a single time point and serum PK data were variable, the distribution of PF-06687234 in the target tissue remains uncertain. While there was no statistically significant difference in the proportion of participants achieving modified clinical remission between the two groups, four out of seven participants achieved an endoscopic improvement in the PF-06687234 plus infliximab group, compared to two out of eight participants in the infliximab-only group. At the group level, the differences in point estimates for both endoscopic improvement and clinical response favor the PF-06687234 plus infliximab group, perhaps suggestive of a trend toward efficacy. Therefore, PF-06687234 may still hold promise for the treatment of UC and our “IL-10 hypothesis” that clinical benefit would result from sustained higher colonic concentrations of IL-10 at the site of intestinal inflammation may be evaluated in future investigations.
This study included some limitations. First, because of the difficulty to recruit participants and the anticipated IA that halted the study for futility, the sample size was small, resulting in limited statistical power (a reluctance among participants to attend weekly study visits may also have limited sample size). However, halting the study for futility protected participants from an ineffective therapy, prioritizing their safety. Second, this study only assessed one dose level of PF-06687234, which may not be optimal due to a potential nonmonotonic clinical effect, especially in a difficult-to-treat population. 42 The complex biological activity of IL-10 could have also contributed to the observed results. This cytokine, known to exhibit both anti-inflammatory and pro-inflammatory effects (producing IFN-γ) at high doses but not at low doses, could display a hormetic response, where its effects vary at different concentrations.17,43 Given the use of single dose level in this study, a possibility of the pro-inflammatory effect balancing out the anti-inflammatory effect at the selected dose cannot be ruled out. Dose selection was further complicated by a lack of prior comprehensive understanding of the PK of the SC formulation of PF-06687234. Concerns regarding the theoretical risks of developing NAbs to endogenous IL-10 and the development of chronic colitis limited prior PK assessment in healthy volunteers. Moreover, analyses were not performed for any biomarkers of target engagement or downstream biologic activity of IL-10, such as phosphorylated signal transducer and activator of transcription 3 (pSTAT3) or IL-10 receptor expression. This further limited our ability to confirm the pharmacological effects of PF-06687234 in humans.
Conclusion
PF-06687234 was well tolerated, with no safety signals of note identified. However, there was no evidence that treatment with PF-06687234 was able to enhance the effectiveness of infliximab (or biosimilar) in improving modified clinical remission when given in combination in this population. Although not statistically significant, we observed a higher proportion of participants with endoscopic remission in the PF-06687234 plus infliximab group compared with the infliximab-only group. Nevertheless, we were unable to confirm the success of our approach to enhance mucosal IL-10 concentration via conjugation to F8, and so the “IL-10 hypothesis” remains not fully tested. The single-dose study design and small sample size may have contributed to these findings. While we did not observe a synergism between PF-06687234 and infliximab, the combination of infliximab with other compounds, and hence other mechanisms of action, may yield important clinical benefits. Thus, there remains an unmet need for further innovative treatment approaches for patients with active UC to enhance and maintain remission of the disease.
Supplemental Material
sj-docx-1-tag-10.1177_17562848251410784 – Supplemental material for Efficacy and safety of the interleukin-10–fragment F8 fusion protein PF-06687234 as add-on therapy to infliximab in patients with active ulcerative colitis: a randomized, phase IIa clinical trial
Supplemental material, sj-docx-1-tag-10.1177_17562848251410784 for Efficacy and safety of the interleukin-10–fragment F8 fusion protein PF-06687234 as add-on therapy to infliximab in patients with active ulcerative colitis: a randomized, phase IIa clinical trial by Stefan Schreiber, Silvio Danese, Séverine Vermeire, Rupert W. Leong, Vivek Pradhan, Ravi Shankar P. Singh, Dahong Yu, Dean Messing, Hendrik Neubert, Joe Palandra, Christopher Tehlirian and Jeremy D. Gale in Therapeutic Advances in Gastroenterology
Supplemental Material
sj-docx-2-tag-10.1177_17562848251410784 – Supplemental material for Efficacy and safety of the interleukin-10–fragment F8 fusion protein PF-06687234 as add-on therapy to infliximab in patients with active ulcerative colitis: a randomized, phase IIa clinical trial
Supplemental material, sj-docx-2-tag-10.1177_17562848251410784 for Efficacy and safety of the interleukin-10–fragment F8 fusion protein PF-06687234 as add-on therapy to infliximab in patients with active ulcerative colitis: a randomized, phase IIa clinical trial by Stefan Schreiber, Silvio Danese, Séverine Vermeire, Rupert W. Leong, Vivek Pradhan, Ravi Shankar P. Singh, Dahong Yu, Dean Messing, Hendrik Neubert, Joe Palandra, Christopher Tehlirian and Jeremy D. Gale in Therapeutic Advances in Gastroenterology
Footnotes
Acknowledgements
The authors would like to thank the participants who volunteered in this study and the investigators at the different clinical sites. The authors would like to acknowledge Dr. Pranab Ghosh for his valuable assistance in establishing the interim analysis criteria for this study. Medical writing support, under the guidance of the authors, was provided by Megan Melody, MSc, and Niall Tyrer, MBiolSci, and was funded by Pfizer, New York, NY, USA, in accordance with Good Publication Practice (GPP 2022) guidelines (Ann Intern Med 2022; 175: 1298–1304).
Declarations
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.