
Abstract
Background:
Vedolizumab is a gut-selective, anti-α4β7 integrin antibody treatment for ulcerative colitis (UC) and Crohn’s disease (CD).
Objectives:
Long-term vedolizumab treatment persistence and safety were evaluated in patients who completed GEMINI LTS (NCT00790933) or VERSIFY (NCT02425111) clinical trials and continued treatment in the open-label vedolizumab extended access program (XAP).
Design:
A phase IIIb/IV clinical study.
Methods:
This phase IIIb/IV, prospective, multinational study (NCT02743806) was conducted from August 2016 to January 2023 in adults with UC/CD treated with vedolizumab 300 mg intravenous every 8/4 weeks (Q8W/Q4W).
Results:
Of 331 (UC, 142; CD, 189) patients enrolled, 311 from GEMINI LTS (UC, 142; CD, 169) and 20 with CD from VERSIFY, most (295/331) started the XAP study on Q8W dosing (UC, 93% (132/142); CD, 86% (163/189)). The mean (SD) disease duration at XAP study baseline was 14 (7) years for UC and 3 (7) years for CD. Vedolizumab Q8W treatment persistence for ⩾6 months without relapse was 98% (129/132) for UC and 95% (154/163) for CD. Twenty-seven patients (9%) were re-escalated from Q8W to Q4W dosing: UC, 8% (11/132); CD, 10% (16/163). The mean (SD) vedolizumab treatment duration in the XAP was 3.5 (1.6) years for UC and 3.6 (1.7) years for CD. In the UC and CD groups, 13% and 16% of patients had serious adverse events (SAEs), respectively (1% per group was treatment related); two (1%) and four (2%) had serious infections, respectively, none related to treatment. One patient died from chronic obstructive pulmonary disease.
Conclusion:
In this prospective longitudinal study, patients had high persistence with vedolizumab Q8W. SAE, relapse, and reescalation to Q4W dosing rates were low.
Trial registration:
The study protocol was registered at ClinicalTrials.gov (https://clinicaltrials.gov/; NCT02743806).
Plain language summary

Introduction
Treatments for inflammatory bowel disease (IBD) can be effective, although careful monitoring is required because some patients fail to achieve durable remission due to primary nonresponse or secondary loss of response to treatment over time.1–5 Failure to optimize treatment outcomes over the long term can lead to uncontrolled symptoms and significant complications, including surgical intervention and reduced patient quality of life.6,7
In IBD, treatment persistence has been suggested to have value as a proxy measure of treatment effectiveness in the real-world setting.8,9 Although treatment persistence can give a reflection of positive patient treatment experiences and physician preferences, 10 low rates of persistence are often synonymous with suboptimal treatment outcomes and can be an indicator of poor tolerability.11,12
Vedolizumab is a gut-selective humanized monoclonal anti-α4β7 integrin antibody that is approved in >70 countries worldwide for the treatment of patients with moderately to severely active Crohn’s disease (CD) or ulcerative colitis (UC).13–15 The efficacy and safety of vedolizumab treatment for patients with moderate to severe CD and UC is well established from clinical studies, including the GEMINI and VERSIFY programs and from real-world clinical experience,16–22 with safety data now available for >1 million patient-years of vedolizumab exposure. 23
Patients who had successfully completed the GEMINI LTS or the VERSIFY study could continue treatment in the vedolizumab extended access program (XAP). GEMINI LTS was a phase III, single-arm, open-label study of patients with moderate to severe CD or UC given vedolizumab 300 mg intravenously every 4 weeks (Q4W) and followed for up to 9 years. 20 VERSIFY was a phase IIIb, open-label, single-arm study of patients with moderate-to-severe CD, of which patients in a VERSIFY substudy were administered vedolizumab 300 mg intravenously every 8 weeks (Q8W) over 52 weeks. 18 The XAP study aimed to assess long-term treatment persistence with Q8W vedolizumab dosing, the occurrence of dose frequency escalation, and the safety of vedolizumab 300 mg in adult patients with UC or CD who rolled over from GEMINI LTS or VERSIFY.
Methods
Patients
The XAP study (NCT02743806; https://clinicaltrials.gov/study/NCT02743806) was a phase IIIb/IV, multinational, multicenter open-label study conducted between December 2016 and January 2023. Institutional Review Board or Ethics Committee approval for the study protocol was obtained at each study site, and all patients provided written informed consent before enrollment. The study was conducted according to the International Council for Harmonization Good Clinical Practice Guidelines, ethical principles that have their origins in the Declaration of Helsinki.24,25 The reporting of this study conforms to the Consolidated Standards of Reporting Trials (CONSORT 2025) statement (available as Supplemental Material). 26 Patients eligible for inclusion were adults with UC or CD who participated in the GEMINI LTS (NCT00790933) 19 or VERSIFY (NCT02425111) 18 clinical trials; specifically, those patients who would continue to benefit from vedolizumab (in the opinion of the investigator) but for whom the drug was not commercially available at the time of patient completion of these two qualifying studies. Clinical remission status was evaluated in patients at enrollment. In patients with UC, clinical remission was defined as a partial Mayo score ⩽2. Clinical remission in patients with CD was defined as Harvey–Bradshaw Index (HBI) score ⩽4 (GEMINI LTS) or CD activity index score ⩽150 (VERSIFY). Results from pharmacokinetic analyses and interim study data from the XAP study have been reported previously.24,25
Study treatment
Following the enrollment visit (final dosing visit of the qualifying study), patients had vedolizumab Q8W dosing visits (patients with UC or CD from GEMINI LTS were to have dose frequency reduction from Q4W to Q8W, and patients with CD from VERSIFY were to be maintained on Q8W dosing). In the XAP study, patients were to receive a 300 mg dose of vedolizumab by intravenous infusion Q8W (±7 days) for 56 weeks or until vedolizumab became available to the patient through commercial channels (including reimbursement) or until patient withdrawal. Dosing regimens at study entry could be varied at the discretion of the study investigator.
Dosing changes
Dosing changes were permitted during the study according to the clinical judgment of the study investigator: if patients experienced reduced efficacy on Q8W dosing, the dosing frequency could be increased to Q4W; Q8W dosing could also be reintroduced during the study at the investigator’s discretion.
Study assessments and endpoints
Treatment persistence
Treatment persistence was assessed in patients starting the XAP study on vedolizumab Q8W dosing and was measured as the proportion of these patients who continued receiving vedolizumab Q8W dosing for ⩾6 months without relapse. Incidence of relapse-free persistence on Q8W dosing for ⩾6 months and time to relapse were also assessed in the study. For these assessments, relapse was defined as any of these events that occurred after XAP study enrollment: (1) vedolizumab dose escalation from Q8W to Q4W; (2) premature vedolizumab treatment or study withdrawal due to loss of response to vedolizumab treatment (reason for discontinuation reported as “no longer an adequate benefit”); or (3) worsening of UC/CD reported as an adverse event (AE; termed “colitis ulcerative,” “Crohn’s disease,” or “proctitis”) fulfilling any of the following criteria: AE of severe intensity; AE of mild or moderate intensity associated with initiation or dose increase of corticosteroids, immunosuppressants/immunomodulators, antitumor necrosis factor alpha (TNFα) agents, or other biologic comedication; AE with associated dose increase of vedolizumab; AE leading to premature discontinuation of the study or of vedolizumab treatment; or AE classified as a serious AE (SAE).
Dose frequency escalation
Incidence of dose escalation from Q8W to Q4W and time to dose escalation were calculated for patients starting the XAP study on Q8W dosing.
Disease relapse
The proportion of patients experiencing a disease relapse event during the study and the time to disease relapse were assessed (relapse events were defined previously for the assessment of treatment persistence).
Treatment compliance
Treatment compliance, or the proportion of planned vedolizumab doses that were administered in the XAP study ((number of vedolizumab doses administered/number of planned doses) × 100), was also assessed. The compliance rate was then summarized by the number and percentage of patients falling into the following categories of compliance: <50%, 50% – <80%, 80%–120%, and >120%.
Safety
Recorded from study enrollment through to the last study visit (or early study termination visit) were AEs, SAEs, AEs of special interest (AESIs; including serious infections, opportunistic infections such as progressive multifocal leukoencephalopathy, malignancies, liver injury, infusion-related hypersensitivity reactions, and injection site reactions), AEs leading to study discontinuation, AEs leading to IBD-related hospitalizations, and vital signs.
Statistical analyses
The assessment of persistence with the vedolizumab Q8W dosing regimen, incidence of relapse, and dose escalation was analyzed for the cohort of patients who were switched to Q8W dosing at XAP study enrollment or who were already receiving Q8W dosing at XAP study enrollment. The safety analysis was performed based on data from the full analysis set, which included all patients receiving at least one dose of vedolizumab during the XAP study. Data were summarized descriptively. All statistical analyses were conducted using SAS version 9.4 (SAS Institute, North Carolina, USA). Kaplan–Meier curves were produced for time to relapse and time to dose escalation (by disease type and the qualifying study). Patients who completed the XAP study or dropped out prior to experiencing the respective event were censored appropriately at their last date within the study (date of study completion or date of premature discontinuation, whichever was applicable).
Results
Patient characteristics
In total, 331 patients were enrolled in the XAP study. Of these, 142 were diagnosed with UC (from GEMINI LTS only) and 189 with CD (169 from GEMINI LTS and 20 from VERSIFY). Patient demographic and disease characteristics at the XAP study baseline are shown in Table 1.
Baseline demographics and clinical characteristics.

Clinical remission in patients with UC was defined as a partial Mayo score ⩽2. Clinical remission in patients with CD was defined as an HBI score ⩽4 (GEMINI) or a CD activity index score ⩽150 (VERSIFY).
HBI score was not available for patients enrolled in VERSIFY.
BMI, body mass index; CD, Crohn’s disease; CS, corticosteroids; HBI, Harvey–Bradshaw Index; IMM, immunosuppressant; Q8W, every 8 weeks; TNF, tumor necrosis factor; UC, ulcerative colitis; XAP, extended access program.
The average time since IBD diagnosis for patients enrolled in the XAP study was approximately 13 years, and >80% of patients were in disease remission at study baseline. Patients who had received prior anti-TNFα treatment made up 20% of the cohort with UC and 39% of patients with CD. For patients in the XAP study, the mean (SD) duration of vedolizumab treatment from the first dose of vedolizumab in a prior study up to XAP study baseline was 6.2 (1.8) years. For patients with UC from GEMINI LTS (n = 142: 107 from GEMINI I (NCT00783718), 24 receiving vedolizumab de novo in GEMINI LTS, and 11 from C13004 (NCT00619489)), the mean (SD) duration of vedolizumab treatment at XAP study baseline was 6.8 (1.3) years. For patients with CD from GEMINI LTS (n = 169: 110 from GEMINI II (NCT00783692), 39 from GEMINI III (NCT01224171), and 20 treated de novo in GEMINI LTS), the mean (SD) duration of vedolizumab treatment at XAP study baseline was 6.3 (1.0) years. The corresponding value for 20 patients with CD from VERSIFY was 0.9 (0.01) years, and the total mean (SD) duration of vedolizumab treatment at XAP baseline for all patients with CD was 5.8 (1.9) years.
Most patients (89.1% (295/331)) started the XAP study on the Q8W dosing regimen (UC, 93.0% (132/142); CD, 86.2% (163/189)). Of the 142 patients from GEMINI LTS with UC, 132 started the XAP study on Q8W dosing. Of 169 patients enrolled from GEMINI LTS with CD, 143 initiated XAP on the Q8W regimen, and 20 patients with CD enrolled from VERSIFY continued Q8W dosing.
The mean duration of vedolizumab treatment in the study was comparable between patients with UC and CD. For patients with UC, the mean (SD) duration of vedolizumab treatment in the XAP study was 3.5 (1.6) years. For patients with CD, the mean (SD) duration of vedolizumab treatment in the XAP study was 3.6 (1.7) years for patients from GEMINI LTS and 3.5 (1.7) years for patients from VERSIFY. Collectively, the mean duration of vedolizumab exposure from the first dose in the qualifying study to the last dose in the XAP study was 9.5 (1.9) or 4.4 (1.6) years for patients who rolled over from GEMINI LTS or VERSIFY, respectively.
Treatment persistence
Among patients with UC and CD initiating the XAP on the Q8W regimen (n = 295), 283 (95.9%) demonstrated persistence with Q8W dosing for ⩾6 months without relapse: 129 of 132 (97.7%) patients with UC and 154 of 163 (94.5%) patients with CD (Figure 1).
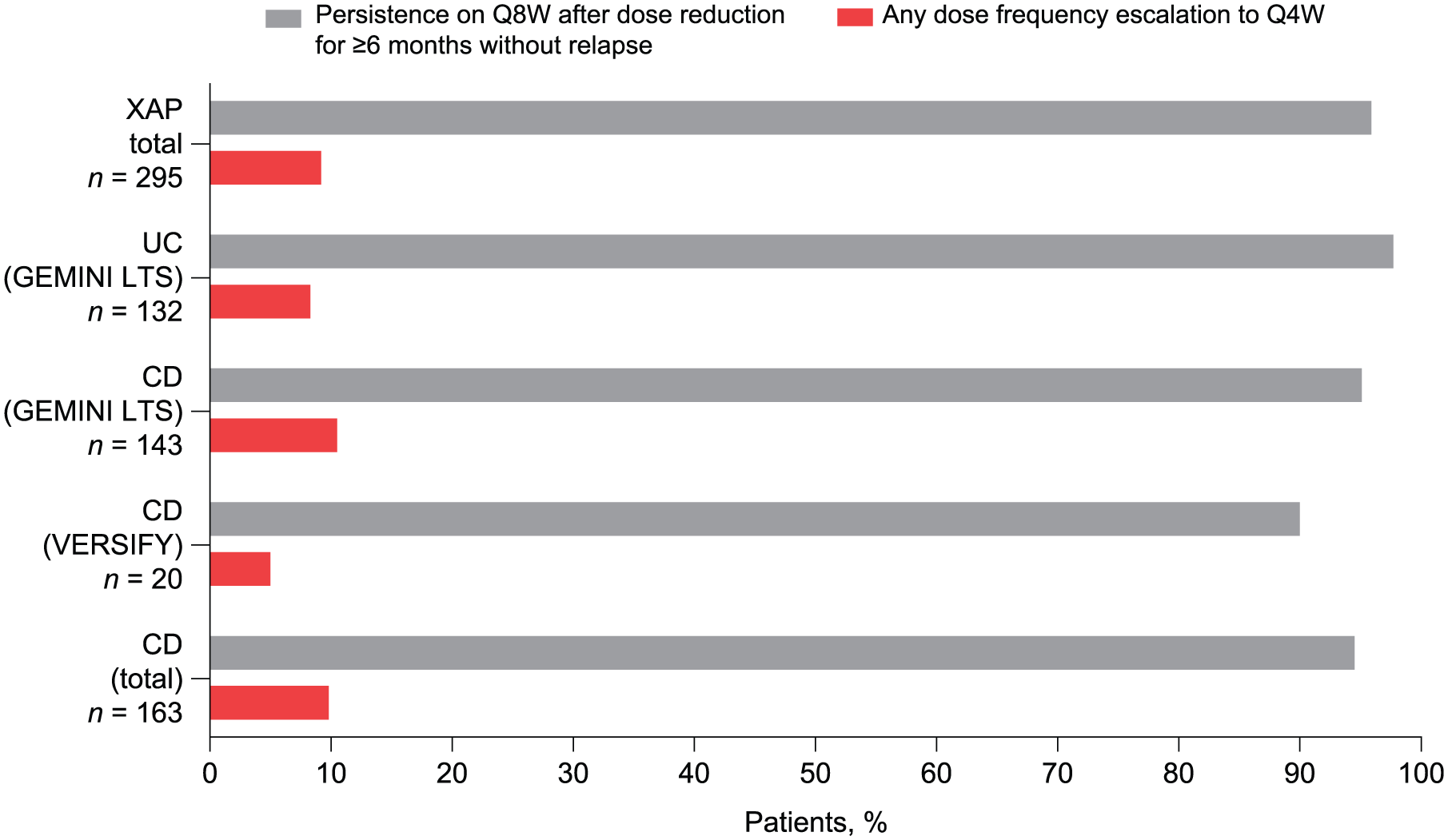
Vedolizumab persistence. Patients with persistence with vedolizumab treatment Q8W for ⩾6 months without relapse and dose escalation to Q4W among patients initiating the XAP study on the Q8W regimen from GEMINI LTS and VERSIFY.
When examining treatment persistence by the qualifying study, for patients from GEMINI LTS, persistence with Q8W dosing after dose reduction for ⩾6 months without relapse was reported for 129 of 132 (97.7%) patients with UC and 136 of 143 (95.1%) patients with CD. In addition, 18 of 20 (90.0%) patients from VERSIFY had persistence for ⩾6 months without relapse with Q8W dosing in the XAP study.
Dose frequency escalation
Only 8.3% (11/132) of patients with UC and 9.8% (16/163) with CD (15 patients from GEMINI LTS and 1 from VERSIFY) who started Q8W dosing in the XAP study had any dose escalation to Q4W during the study. The median (range) time from last dose in the qualifying study to dose escalation in the XAP study was 540 (211–1108) days for patients with UC and 423.5 (85–1908) days for patients with CD. Kaplan–Meier curves for time to dose escalation for patients who started the XAP study on Q8W dosing from GEMINI LTS are shown in Figure 2.

Kaplan–Meier curves for time to first dose frequency escalation from Q8W to Q4W for patients with (a) ulcerative colitis and (b) Crohn’s disease who started the extended access program from GEMINI LTS on Q8W dosing. T0 was the time of the last dose of vedolizumab in GEMINI LTS.
Disease relapse
There were 46 of 295 (15.6%) patients with any relapse event in the XAP study (note, dose escalation was included in the definition of relapse event). This included 16 of 132 (12.1%) patients with UC and 30 of 163 (18.4%) patients with CD (28/143 (19.6%) patients from GEMINI LTS and 2/20 (10.0%) from VERSIFY; Figure 3). The median (range) time to relapse was 523 (140–1466) days in patients with UC and 439.5 (22–1967) days in patients with CD. The Kaplan–Meier curves for time to first relapse for patients from the GEMINI LTS study who started the XAP on Q8W dosing are shown in Figure 4.
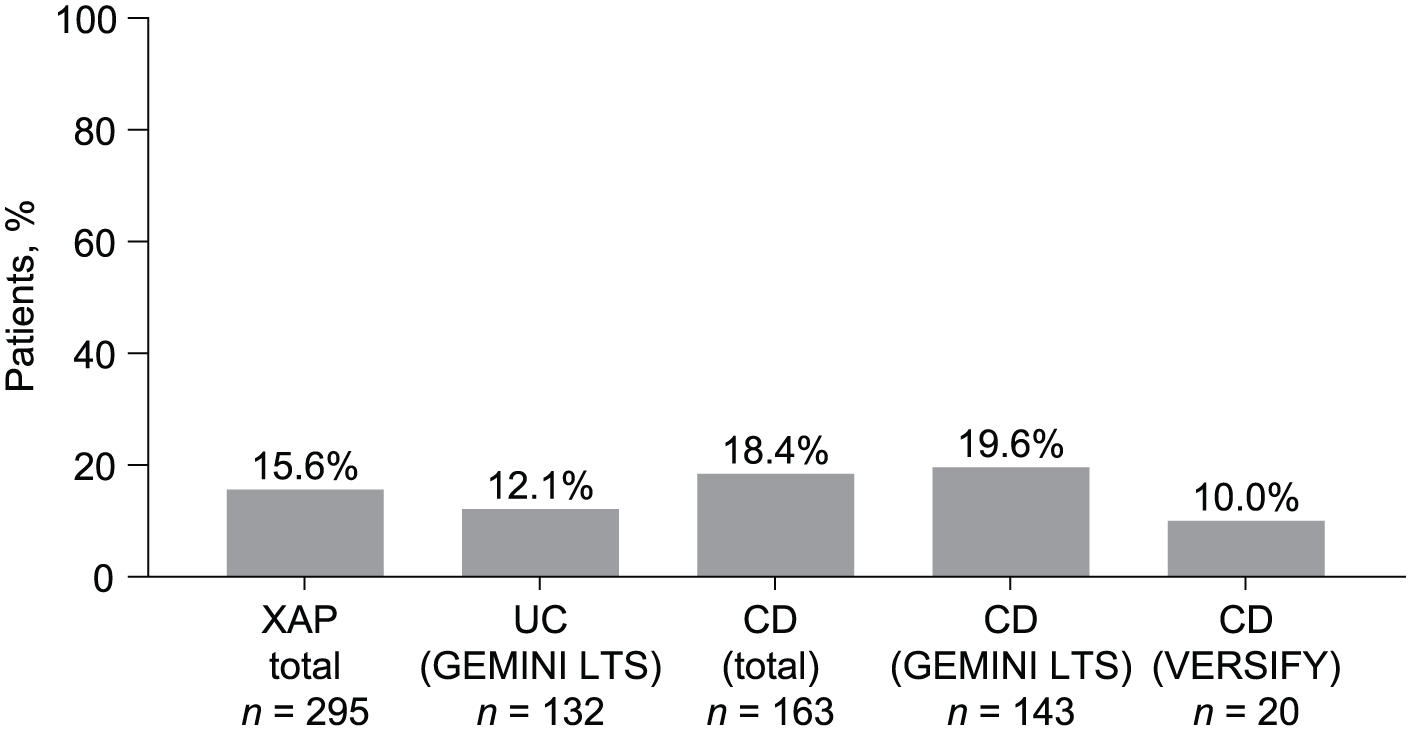
Proportion of patients with relapse. Relapse definition: any of these events occurring after XAP enrollment: (1) vedolizumab dose escalation from every 8 weeks to every 4 weeks; (2) premature withdrawal from study drug or study owing to loss of response to study treatment; and (3) worsening of UC/CD reported as an adverse event.

Kaplan–Meier curves for time to first relapse for patients who started the extended access program from GEMINI LTS on Q8W dosing: (a) patients with ulcerative colitis and (b) patients with Crohn’s disease. T0 was the time of the last dose of vedolizumab in the qualifying study (GEMINI LTS).
Vedolizumab treatment compliance
Treatment compliance for the duration of the XAP study was recorded in the 80%–120% compliance category for the majority of patients: 133 of 142 (93.7%) with UC and 185 of 189 (98.9%) with CD. There were no differences in compliance for patients who deescalated to Q8W dosing versus those who were stable with Q8W at study entry.
Adverse events
A total of 205 of 331 (61.9%) patients had AEs during the XAP study. AEs occurred in 87 of 142 (61.3%) patients with UC and 118 of 189 (62.4%) patients with CD after enrollment in the XAP study and were related to vedolizumab treatment in 3 of 142 (2.1%) and 8 of 189 (4.2%) patients with UC and CD, respectively. The majority of AEs were mild or moderate in severity (Table 2).
Overview of AEs and SAEs in the XAP.

The summary included AEs that occurred from the time the patient received the last dose in the qualifying study through the end of the study visit. Patients who experienced more than one AE were counted a single time at the closest relationship level (related > not related) and a single time at maximum intensity level (severe > moderate > mild). Percentages were based on the total number of patients in the full analysis set for each group.
AE, adverse event; CD, Crohn’s disease; IBD, inflammatory bowel disease; SAE, serious adverse event; UC, ulcerative colitis; XAP, extended access program.
AEs leading to study drug discontinuation occurred in 6 of 142 (4.2%) patients with UC and 3 of 189 (1.6%) patients with CD. These were related to treatment in 1 of 142 (0.7%) patients with UC and 1 of 189 (0.5%) patients with CD. A total of 5 of 142 (3.5%) patients with UC and 14 of 189 (7.4%) patients with CD experienced an AE that led to IBD-related hospitalization; none of these AEs were considered to be treatment-related.
A summary of the type of AEs that occurred in ⩾5% of patients with UC and CD is shown in Supplemental Table 1. In patients with UC, the most common AEs were worsening UC (20 patients), hypertension (12 patients), and coronavirus (COVID-19) infection (11 patients). In patients with CD, the most common AEs were CD worsening (33 patients) and nasopharyngitis and arthralgia (10 patients each).
Serious adverse events
A total of 50 of 331 patients (15.1%) reported at least one SAE; 13.4% (19/142) in patients with UC and 16.4% (31/189) in patients with CD. Overall, the most frequently reported SAEs occurring in ⩾1% of patients in the XAP study were worsening of CD (five patients (3.0%)), then worsening of UC (four patients (2.8%); Table 3). Most of the SAEs reflected worsening of the underlying disease and were moderate or mild in intensity. Overall, only 21 of 331 patients (6.3%) experienced SAEs that were assessed by the investigator as severe in intensity.
Summary of SAEs that occurred in ⩾1% of patients.

The summary included AEs that occurred from the patients’ last dose in the qualifying study through the end of the study visit. If the patient reported at least one event, they were counted only once at each level of summarization. SAEs were sorted in descending order of preferred term incidence based on the extended access program total. The percentage was based on the number of patients in the full analysis set for each subgroup. Medical Dictionary for Regulatory Activities (version 25.0) was used for coding AEs.
AE, adverse event; CD, Crohn’s disease; SAE, serious adverse event; UC, ulcerative colitis.
Aside from worsening of UC, the most frequently reported SAEs in patients with UC were joint dislocation (two patients) and malignant colorectal neoplasms (two patients). For severe SAEs in the UC cohort, 10 patients experienced 11 SAEs that were severe in intensity, including 4 of UC worsening, and 1 each of large intestine perforation, coronavirus (COVID-19) pneumonia, joint dislocation, osteoarthritis, pseudopolyposis, colon adenocarcinoma, and liver metastases.
Aside from worsening of CD, the most frequently reported SAEs in patients with CD were myocardial infarction (two patients) and subileus (two patients). For severe SAEs in patients with CD, 11 patients experienced 13 severe SAEs, which included 2 severe SAEs of myocardial infarction, 2 of CD worsening, and 1 each of nausea, vomiting, abdominal abscess, chronic obstructive pulmonary disease, anemia, large intestinal obstruction, central nervous system metastases, malignant lung neoplasm, and inguinal hernia.
Overall, two patients (0.6%) experienced SAEs reported by the investigator as being related to study treatment; these were adenocarcinoma of the colon and liver metastases in a patient with UC and spontaneous abortion in a patient with CD.
Serious infections
In total, 6 of 331 patients (1.8%) had 6 infection events classified as SAEs in the study; none were considered related to vedolizumab treatment. For patients with UC, two patients (1.4%) had coronavirus (COVID-19) infections. The serious infections reported in four patients with CD (2.4%) were pneumonia, viral gastrointestinal infection, anal abscess, and abdominal abscess (one patient for each).
AEs of special interest
Apart from serious infections, other AESIs reported among patients with UC included squamous cell carcinoma of the skin (two AESIs in one patient), pseudopolyposis, colon cancer, colorectal adenocarcinoma, colon adenocarcinoma, and metastases to the liver (one patient for each). Two AESIs, colon adenocarcinoma and liver metastases, were assessed by the investigator as being related to vedolizumab treatment.
Among patients with CD, other AESIs aside from serious infections included one AESI of asthenia in one patient (assessed by the investigator as being related to vedolizumab treatment). Four AESIs reported in two patients were metastases to the central nervous system, malignant lung neoplasm, malignant melanoma in situ, and squamous cell carcinoma of the skin. One AESI event of anaphylaxis was reported in one patient.
Deaths
One death was reported during the study and was considered unrelated to vedolizumab. The event was attributed to the exacerbation of chronic obstructive pulmonary disease, which was a preexisting condition.
Other safety parameters
There were no changes of note in vital signs or physical examination results across the XAP study visits. There were five patients with CD from the GEMINI LTS study and one patient with UC from the GEMINI LTS study who discontinued vedolizumab treatment due to pregnancy. One incidence of pregnancy with an outcome of “abortion spontaneous” was reported as an SAE related to vedolizumab treatment in a patient with CD from the GEMINI LTS study.
Overall, the safety data were in line with the safety profile of vedolizumab previously reported and raised no new safety concerns.
Discussion
In this longitudinal, prospective, open-label study, high rates of treatment persistence were reported with vedolizumab Q8W treatment in patients who had successfully completed the GEMINI LTS or VERSIFY clinical studies; 98% of patients with UC and 95% with CD demonstrated persistence with Q8W vedolizumab dosing for ⩾6 months without relapse, while <10% changed to Q4W dosing. Furthermore, high treatment persistence was observed both in patients previously on long-term vedolizumab who reduced their treatment frequency by 50% from Q4W to Q8W at enrollment (i.e., those from GEMINI LTS; UC, 97.7%; CD, 95.5%) as well as in patients with less overall prior experience on vedolizumab Q8W (i.e., those from VERSIFY; 90.0%). Only 12% of patients with UC and 18% of patients with CD had disease relapse during the XAP study. High patient persistence and low rates of Q4W dose frequency reescalation and relapse were documented during a median of 4 years of patient follow-up.
The data on safety parameters reported in patients receiving vedolizumab treatment during this study were in line with the safety profile of vedolizumab reported previously. There were no new trends or safety issues identified. Most of the AEs were assessed as being unrelated to vedolizumab and were mild to moderate in intensity. SAEs were reported in 15% of patients during the study, which most commonly reflected worsening of the underlying disease (CD, 3.0%; UC, 2.8%) and were moderate or mild in intensity; only 6% of patients had severe SAEs and only two patients (0.6%) had SAEs considered related to vedolizumab treatment. Serious infections occurred in six patients (1.8%), of which none were considered related to vedolizumab treatment.
Persistence data from patients who continued vedolizumab treatment after first receiving vedolizumab in a clinical trial also confirm previous results obtained with vedolizumab in the real-world setting. Demuth et al. 27 published a systematic literature review and meta-analysis on the persistence with vedolizumab treatment in patients with IBD. In this analysis of real-world data, approximately three-quarters of patients with UC and nearly two-thirds of patients with CD persisted with vedolizumab treatment at 12 months, despite being a largely biologic-refractory patient population. Higher vedolizumab treatment persistence was observed in those patients naïve to biologic therapy versus biologic-experienced patients. 27 The EVOLVE 28 retrospective chart review study of biologic therapy received first line in biologic-naïve patients with UC and CD showed that vedolizumab and anti-TNFα treatments had equal effectiveness in terms of disease symptom control. In the EVOLVE chart review, rates of treatment persistence by 24 months were higher in patients with UC who had been treated with vedolizumab versus anti-TNFα agents, and incidence rates of SAEs and serious infections were significantly lower in patients treated with vedolizumab than in those treated with anti-TNFα agents. A comparison of infliximab and vedolizumab as first-biologic treatments in a real-world cohort study of 420 patients from Australia with moderate to severe UC reported persistence with vedolizumab of >50.2 months versus 22.2 months with infliximab (p = 0.001). 29 A retrospective cohort study of 217 biologic-naïve patients with UC from a tertiary center in Israel similarly reported vedolizumab treatment persistence of 265.6 weeks, which was significantly longer than treatment persistence for infliximab of 106.5 weeks (p = 0.001), including when the analysis was adjusted for factors associated with disease severity. 30
The main limitations of the XAP study relate to its design; although the study was prospective, treatment was open-label. There was potential for selection bias because study entry was restricted to patients who had experienced a benefit with vedolizumab treatment in a previous clinical trial. Also, patients eligible for the XAP study were those without ongoing access to vedolizumab treatment outside of a clinical trial setting, increasing the likelihood of selection bias related to barriers to vedolizumab treatment access at that time. Patients fulfilling eligibility criteria for the qualifying GEMINI LTS and VERSIFY studies may not be completely representative of the general population of patients with IBD. In addition, there were no fixed dosing intervals imposed at treatment initiation as in other clinical studies; not all patients initiated Q8W dosing in the XAP study because dosing frequency at study initiation could be changed at the discretion of the study investigator. Likewise, the decision of the investigator to increase dosing frequency during the study did not require confirmation of disease worsening by objective assessment, such as endoscopy or biomarker measurement, and could have impacted the observed treatment persistence.
Conclusion
Patients had high rates of persistence with vedolizumab Q8W treatment during a median of 4 years of follow-up in the XAP long-term, prospective study, even in patients with dosing frequency reduced from Q4W to Q8W at study entry. Rates of disease relapse and dose frequency reescalation during the study were low for patients receiving vedolizumab Q8W. These findings lend further support to the durability of vedolizumab treatment effectiveness, even at a reduced dosing frequency, in addition to the long-term safety of vedolizumab treatment in patients with IBD.
Supplemental Material
sj-docx-1-tag-10.1177_17562848251406092 – Supplemental material for Final results from the vedolizumab extended access program multinational study demonstrate long-term treatment persistence and safety
Supplemental material, sj-docx-1-tag-10.1177_17562848251406092 for Final results from the vedolizumab extended access program multinational study demonstrate long-term treatment persistence and safety by Silvio Danese, Milan Lukáš, Miroslava Volfová, Grażyna Rydzewska, Shashi Adsul, Dirk Lindner, Stephen Jones and Séverine Vermeire in Therapeutic Advances in Gastroenterology
Supplemental Material
sj-pdf-2-tag-10.1177_17562848251406092 – Supplemental material for Final results from the vedolizumab extended access program multinational study demonstrate long-term treatment persistence and safety
Supplemental material, sj-pdf-2-tag-10.1177_17562848251406092 for Final results from the vedolizumab extended access program multinational study demonstrate long-term treatment persistence and safety by Silvio Danese, Milan Lukáš, Miroslava Volfová, Grażyna Rydzewska, Shashi Adsul, Dirk Lindner, Stephen Jones and Séverine Vermeire in Therapeutic Advances in Gastroenterology
Supplemental Material
sj-pdf-3-tag-10.1177_17562848251406092 – Supplemental material for Final results from the vedolizumab extended access program multinational study demonstrate long-term treatment persistence and safety
Supplemental material, sj-pdf-3-tag-10.1177_17562848251406092 for Final results from the vedolizumab extended access program multinational study demonstrate long-term treatment persistence and safety by Silvio Danese, Milan Lukáš, Miroslava Volfová, Grażyna Rydzewska, Shashi Adsul, Dirk Lindner, Stephen Jones and Séverine Vermeire in Therapeutic Advances in Gastroenterology
Footnotes
Acknowledgements
The study was funded by Takeda Development Center Americas, Inc. The authors would like to thank the patients and their caregivers who participated in this study. Medical writing assistance was provided by Isobel Lever, PhD, and Chase Hellmer, PhD, of Envision Catalyst, an Envision Medical Communications agency, a part of Envision Pharma Group, and was funded by Takeda Development Center Americas, Inc.
Author’s note
Shashi Adsul (Takeda Pharmaceuticals USA, Inc.): At the time the study was conducted and the manuscript was developed.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.