
Abstract
Background:
Fecal microbiota, live-jslm (RBL) is approved in the United States and Canada for prevention of recurrent Clostridioides difficile infection (rCDI) in adults following standard-of-care (SOC) antibiotic treatment.
Objectives:
Provide an updated integrated safety analysis, incorporating final safety data from Punch CD3-OLS.
Design:
Safety data were combined from five RBL trials: three phase II and two phase III trials.
Methods:
Adult participants had documented rCDI and completed SOC therapy before receiving one or two doses of RBL or placebo, rectally administered as one treatment course. Treatment-emergent adverse events (TEAEs) were recorded for ⩽6 months.
Results:
TEAEs were reported in 70.9% (845/1192) of RBL recipients; most TEAEs were mild to moderate and gastrointestinal in nature. Most serious TEAEs were related to preexisting conditions or CDI. There was no clustering of serious TEAEs. Most TEAEs leading to death were related to preexisting conditions.
Conclusion:
Overall, data demonstrate RBL has a favorable 6-month safety profile.
Trial registration:
ClinicalTrials.gov: NCT01925417; NCT02299570; NCT02589847; NCT03244644; NCT03931941.
Introduction
Clostridioides difficile infection (CDI) affects an estimated half a million patients every year in the United States (US), 1 and is recognized as a leading contributor to healthcare-associated infections worldwide. 2 Ongoing high incidence of recurrent CDI (rCDI), defined as CDI that reoccurs within 8 weeks after successful treatment of an initial episode, is considered a major public health challenge.1,3 After the initial episode of CDI, up to 35% of patients will experience recurrence and, of those, up to 60% will experience subsequent episodes of rCDI.1,2 In the United States, between 75,000 and 175,000 cases of CDI each year are due to rCDI. 1
Antibiotic therapy is the current standard-of-care (SOC) treatment for CDI and effectively controls the acute infection by eradicating the vegetative phase.4,5 Patients remain susceptible to recurrence due to persistent dysbiosis; remaining Clostridioides difficile spores after SOC treatment can transition from a dormant state to a metabolically active state (vegetative phase), leading to rCDI.4,5 Microbiota-based products have shown efficacy in preventing rCDI and restoring the composition and diversity of the gut microbiome. 6
Fecal microbiota, live-jslm (REBYOTA®, abbreviated as RBL, previously RBX2660) was approved by the US Food and Drug Administration (FDA) in November 2022 as the first single-dose microbiota-based product for the prevention of rCDI in adults following SOC antibiotic treatment.7,8 In March 2025, RBL received approval from Health Canada for the prevention of rCDI in adults following SOC antibiotic treatment.9,10 During clinical development of RBL, no major safety signals were observed. 11 Although a previous integrated summary of safety data from the prospective RBL trials was published by Lee et al. 11 in 2023, it only included ad hoc data from PUNCH CD3-OLS, the largest trial of a microbiota-based product to date. PUNCH CD3-OLS permitted enrollment of patients with complex comorbidities (e.g., inflammatory bowel disease, mild to moderate immunocompromise) that were excluded from earlier trials, including randomized controlled trials. 12 This manuscript reports an updated analysis incorporating the final safety data from PUNCH CD3-OLS, summarizing trial data over an approximately 10-year period.
Methods
Trial design
Safety data from five completed prospective clinical trials were combined; three phase II trials (PUNCH CD [NCT01925417], PUNCH CD2 [NCT02299570], and PUNCH Open-Label [NCT02589847]) as well as two phase III trials (PUNCH CD3 [NCT03244644] and PUNCH CD3-OLS [NCT03931941]).6,12–16 All trials followed similar protocols. RBL is manufactured using standardized procedures and pathogen screening in collaboration with the FDA to ensure product safety. All trial protocols were approved by the relevant institutional review boards, and were conducted in accordance with the ethical principles of the Declaration of Helsinki, Good Clinical Practice guidelines, and requirements of publicly registered clinical trials. Written informed consent was obtained for all participants prior to trial commencement.
Trials enrolled adults 18 years of age and older with rCDI who received antibiotics for their enrolling CDI episode before RBL administration. Depending on the trial design, the dosing regimen was one or two doses of RBL and/or placebo administered 7 ± 2 days apart; the two-dose regimen was considered one treatment course (Supplementary Figure 1). Four trials (PUNCH CD, PUNCH CD2, PUNCH CD3, and PUNCH CD3-OLS) allowed an unblinded second course of RBL if a CDI recurrence was confirmed within 8 weeks of the first course. By offering this unblinded second course, participants originally administered placebo could have received unblinded RBL and, for the purpose of this analysis, are considered in the any RBL group and not the placebo only group. Trials included up to 6 months of follow-up after the last dose of RBL or placebo; the follow-up schedule restarted after receipt of an unblinded second course, to allow for up to 6 months of follow-up. Two trials (PUNCH CD2 and PUNCH Open-Label) included 24 months of follow-up after the last treatment course, which has been previously published.11,13–15 All trials included stopping rules to allow for medical monitor and data safety monitoring board (DSMB) evaluation of adverse events (AEs) meeting the criteria (e.g., relationship of new pathogenic intestinal infection) and determine if a trial pause or termination was warranted.
Safety analysis
The safety population was defined as any participant who received RBL or placebo; participants who received one dose of RBL were denoted as the RBL (one-dose) group and participants who received one or more doses of RBL were denoted as the any RBL group (Supplemental Figure 1). Safety analyses were presented from baseline to 6 months after the last RBL dose or placebo administration. If a participant received an unblinded second course of RBL, the follow-up schedule was restarted to allow for 6 months of follow-up. Definitions of AEs, treatment-emergent adverse events (TEAEs), and serious TEAEs have been previously published in Lee et al. 11 TEAEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA), version 20.0. TEAEs are presented by MedDRA system organ class (type) and preferred term (exact description of medical condition).
Statistical analysis
Descriptive statistics were reported. A relative risk assessment was used to assess the risk of TEAEs by system organ class and preferred term for participants who received one dose of RBL and participants who received placebo.
Results
Participants
Demographics and baseline characteristics of all participants in the safety population are detailed in Supplemental Table 1. In total, 1192 participants received at least one dose of RBL (any RBL group), 769 participants received one dose of RBL, and 83 participants received placebo only. In the RBL (one dose), placebo only, and any RBL groups, 52.9%, 62.7%, and 49.9% of participants, respectively, were under 65 years of age. Across all treatment groups, most participants were female and White and had experienced at least three episodes (i.e., at least two recurrences) of CDI before enrollment; vancomycin was the most commonly used antibiotic for the enrolling episode. A greater percentage of participants in the any-RBL group had at least four CDI episodes before trial entry compared with the placebo only group (Supplemental Table 1). Regarding medical history, a small percentage of participants had a history of food allergies or celiac disease in the any RBL group; no participants in the placebo only group had a history of food allergies or celiac disease (Supplemental Table 2). In PUNCH CD3-OLS, 74 participants had inflammatory bowel disease, 141 participants had mild to moderate immunocompromising conditions, and 90 participants had irritable bowel syndrome.17–19
Safety
Across all treatment groups, most participants experienced TEAEs that were mild or moderate in severity (Table 1). The most common TEAEs across all groups were in the gastrointestinal disorders system organ class; there was no evidence of increased risk of gastrointestinal TEAEs between the RBL (one dose) and placebo only groups (Figure 1). Most gastrointestinal TEAEs occurred within 2 weeks of placebo or RBL administration in all groups and decreased thereafter (Supplemental Figure 2). Most serious TEAEs were assessed by the investigator as related to a preexisting condition or CDI (Table 1). No events were assessed as only related to RBL or its administration, and a final safety review conducted by the DSMB and FDA determined there were no safety trends of concerns for RBL or its administration. There was no clustering of terms or types of serious TEAEs. No participants with food allergies or celiac disease experienced serious allergic reactions (e.g., anaphylaxis, urticaria, or worsening disease). TEAEs leading to death varied across system organ classes, suggesting no biological correlation (Supplemental Table 3); most were related to preexisting conditions and were unrelated to RBL or its administration. No deaths were assessed to be related to RBL following publication of the initial integrated analysis and the completion of PUNCH CD3-OLS.
Summary of TEAEs within 6 months follow-up of RBL or placebo administration across five RBL clinical trials (safety population).

Data are reported as n (%).
Relatedness classifications are not mutually exclusive and events may have had multiple classifications; events considered possibly related to RBL were not exclusive and could have also been considered related to rCDI and/or preexisting conditions.
Leukocytosis (n = 1), atrial fibrillation (n = 1), abdominal pain (n = 1), ulcerative colitis (n = 1), constipation (n = 1), diarrhea (n = 1), ileus (n = 1), pyrexia (n = 1), rCDI (n = 4), and recurrent acute myeloid leukemia (n = 1).
Leukocytosis (n = 1), atrial fibrillation (n = 1), ileus (n = 1), pyrexia (n = 1), and rCDI (n = 2).
TEAEs leading to death are summarized in Supplemental Table 3.
CDI, Clostridioides difficile infection; RBL, fecal microbiota, live-jslm; rCDI, recurrent CDI; TEAE, treatment-emergent adverse event.
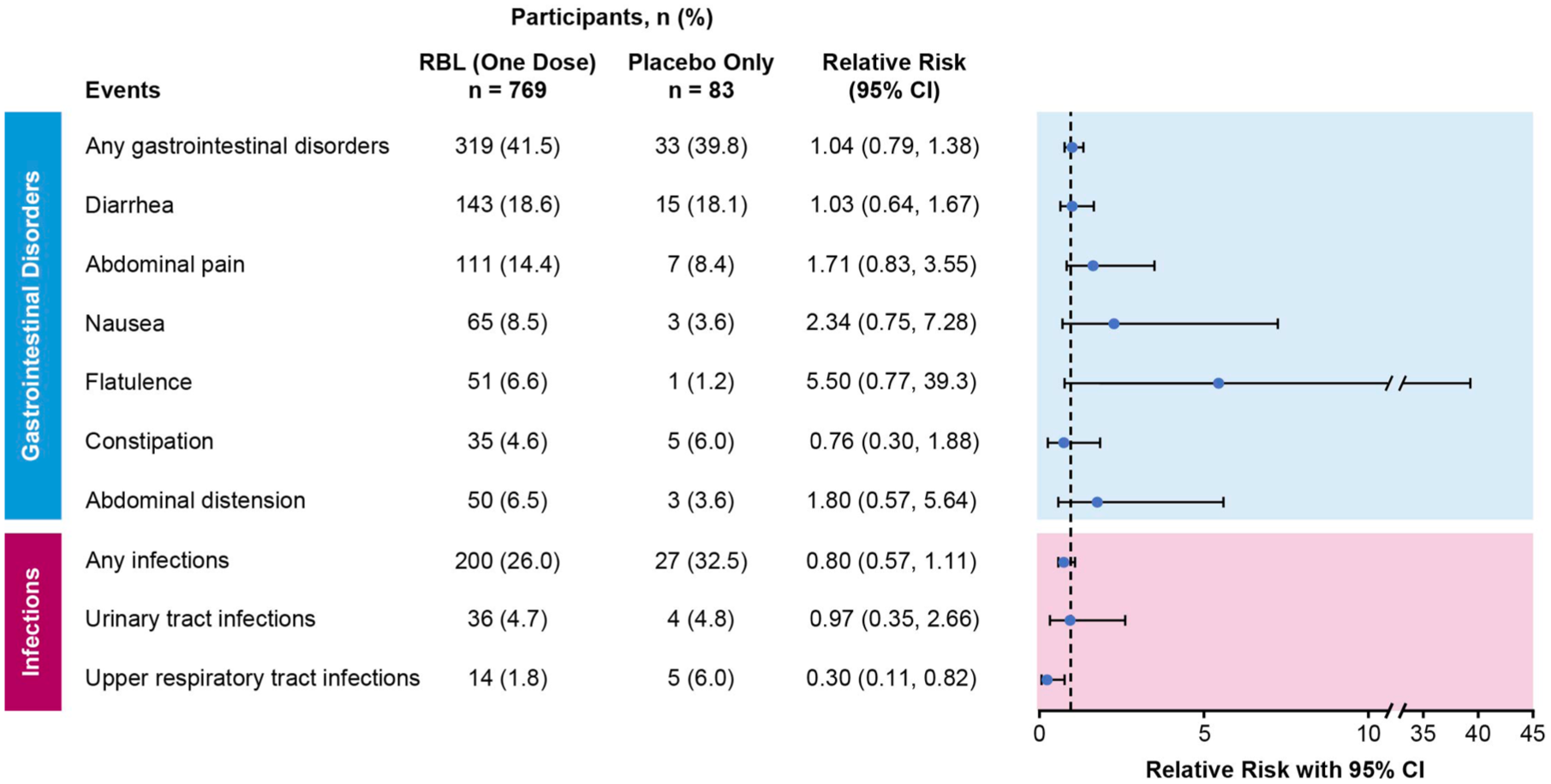
Relative risks and associated 95% confidence intervals of TEAEs by system organ class and preferred term (safety population). TEAEs are reported with an incidence of ⩾5% within 6 months follow-up of RBL or placebo administration across five RBL clinical trials. Relative risk is the ratio of the probability of a participant experiencing a TEAE in the RBL (one dose) group versus the placebo only group. Coding was based on MedDRA Version 20.0.
Discussion
The RBL safety data reported here comprise final data from five completed trials and are updated from the initial integrated analysis in 2023, 11 following the completion of PUNCH CD3-OLS. Safety data from over 1100 RBL recipients through 6 months were combined, including participants from PUNCH CD3-OLS with complex comorbidities (inflammatory bowel disease and mild to moderate immunocompromising conditions). This analysis is the largest safety evaluation of any microbiota-based product to date and further demonstrates RBL is well-tolerated for the prevention of rCDI in adults following SOC antibiotic treatment.
Most TEAEs were mild to moderate in severity and occurred shortly after RBL or placebo administration. Although gastrointestinal TEAEs were commonly reported throughout the clinical development program, there was no evidence of increased risk of these events observed with the RBL (one dose) group versus placebo only group. No new serious safety signals emerged from this analysis. Since the publication of the initial integrated analysis, 11 three new deaths were reported, all of which were related to preexisting conditions and unrelated to RBL or its administration. TEAEs of sepsis and bacteremia leading to death were determined via the safety review process to be unrelated to RBL.
The potential risk of adverse reactions due to food allergens in RBL is unknown 8 and has not been previously reported. Throughout the RBL clinical development program, no dietary restrictions were imposed on donors or trial participants. No RBL recipients with a medical history of food allergies or celiac disease experienced a serious allergic reaction during their trial participation. Additionally, RBL recipients without a medical history of food allergies or celiac disease did not develop any new severe allergic reactions or conditions.
This analysis is limited in that AEs were captured comprehensively and not restricted to product relatedness. Some events, including gastrointestinal TEAEs, may have been related to rCDI and/or preexisting conditions and not RBL or placebo. Additionally, patients with fulminant CDI and a life expectancy of less than 6 months were excluded from all RBL trials. Due to the lack of diversity in the participant populations enrolled across the five trials, AEs applicable to non-White populations may not have been detected.
While inclusion of patients with inflammatory bowel disease and immunocompromising conditions was limited to PUNCH CD3-OLS, safety findings are largely consistent with those in the broader trial population of patients with rCDI, and no new signals were observed.17–19 In PUNCH CD3-OLS, TEAEs were reported in 45.9%, 44.7%, and 57.8% of participants with inflammatory bowel disease, immunocompromising conditions, and irritable bowel syndrome, respectively; serious TEAEs were reported in 1.4%, 4.3%, and 1.1% of participants in these subgroups, respectively (Supplemental Table 4).17–19 The 2024 American Gastroenterological Association clinical practice guidelines suggest use of conventional fecal microbiota transplantation for rCDI in patients who are mildly or moderately immunocompromised, while noting that evidence was insufficient to recommend FDA-approved microbiota-based products due to limited data available as of the review end date of March 2023. 20 Evidence from participants with mild to moderate immunocompromising conditions in PUNCH CD3-OLS, published in 2025, supports the use of RBL in this participant subgroup. 18
Conclusion
Integrated data across all five prospective clinical trials included in this analysis demonstrate RBL is safe and well tolerated in adults with rCDI. This updated integrated safety analysis of RBL is the largest evaluation of any microbiota-based product for the prevention of rCDI following SOC antibiotics reported to date.
Supplemental Material
sj-docx-1-tag-10.1177_17562848251395566 – Supplemental material for Integrated analysis of the safety of fecal microbiota, live-jslm in adults with recurrent Clostridioides difficile infection from five prospective clinical trials: an update
Supplemental material, sj-docx-1-tag-10.1177_17562848251395566 for Integrated analysis of the safety of fecal microbiota, live-jslm in adults with recurrent Clostridioides difficile infection from five prospective clinical trials: an update by Christine Lee, Paul Feuerstadt, Thomas Louie, Lindy Bancke, Beth Guthmueller, Adam Harvey, Frederikke Hoeyer, Robert Orenstein, Erik R. Dubberke and Sahil Khanna in Therapeutic Advances in Gastroenterology
Footnotes
Acknowledgements
The authors thank all the participants and their families and caregivers as well as the investigators and site staff. Medical writing support, under the guidance of the authors, was provided by Megan Payne MChem and Michelle Boland, PhD (ApotheCom; Yardley, PA, USA), and was funded by Ferring Pharmaceuticals, Parsippany, NJ, USA.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.