
Abstract
Background:
Managing patients with primary biliary cholangitis (PBC) who demonstrate an inadequate response to ursodeoxycholic acid or experience intolerable side effects remains a significant clinical challenge.
Objectives:
This study aims to investigate the efficacy and safety of peroxisome proliferator-activated receptor (PPAR) agonists in the treatment of PBC.
Design:
Meta-analysis and systematic review.
Methods:
A systematic search of publications in PubMed, Embase, Web of Science, and the Cochrane Central Register of Controlled Trials was performed. Randomized controlled trials published in English that involved the treatment of PPAR agonists and reported on the levels of alkaline phosphatase (ALP), biochemical response rates, pruritus score, or severe and serious adverse events (AEs) were selected. The primary outcomes assessed were the effects of PPAR agonists on ALP levels and biochemical response rates. Secondary outcomes included the rates of severe or serious AEs and relief of pruritus.
Results:
Fourteen studies with 1137 patients were included. Compared to the control group, PPAR agonists significantly reduced ALP levels by a mean difference of −155.87 U/L (95% confidence interval (CI): −208.30 to −103.44; random-effects). Patients who received PPAR agonists showed a significantly higher biochemical response rate (risk ratio (RR), 4.42; 95% CI: 2.37–8.26; random-effects). Furthermore, there was no significant difference in the rate of severe (RR, 1.05; 95% CI, 0.49–2.28) or serious AEs (RR, 1.02; 95% CI, 0.65–1.60) between the PPAR agonists and placebo groups.
Conclusion:
PPAR agonists are effective and safe to treat patients with PBC.
PROSPERO trial registration:
CRD42024545743.
Introduction
Primary biliary cholangitis (PBC) is an autoimmune disease characterized by immune-mediated injury targeting small bile ducts, which can potentially lead to liver cirrhosis and failure.1,2 Chronic inflammation and granulomatous destruction of small interlobular bile ductules are hallmark features of PBC. 3 A meta-analysis comprising 47 population-based studies shows that the pooled global incidence and prevalence of PBC at 1.76 and 14.60 per 100,000 persons, respectively. 4 Notably, the incidence and prevalence of PBC vary across different regions, being lower in the Asia-Pacific region compared to North America and Europe. 5
Various factors, including specific serum autoantibodies, immune-mediated cellular injury, and genetic (HLA and non-HLA) risk factors, contribute to the development of PBC. Without intervention, this condition can lead to cirrhosis or liver failure. Current treatment primarily targets cholestatic consequences. 6 Ursodeoxycholic acid (UDCA) is the first-line treatment choice for patients with PBC due to its ability to normalize abnormal serum liver tests and improve prognostic surrogate markers. 7 Mechanically, UDCA promotes intracellular and canalicular transport of hydrophobic bile acids, stabilizes hepatocyte membranes, reduces rates of apoptosis of biliary epithelial cells, and mitigates T-cell reactivity by diminishing the expression of adhesion molecules. 8 Nevertheless, up to 40% of PBC patients exhibit an inadequate response to UDCA, and 3%–5% experience unacceptable adverse events (AEs). 9 Furthermore, pruritus significantly affects the quality of life in these patients, yet strong evidence supporting UDCA’s ability to improve pruritus is lacking. 10 Since 2016, obeticholic acid (OCA) has been approved by the FDA for treating PBC patients who have an inadequate response or intolerance to UDCA 11 ; however, recent restrictions on OCA use have been implemented due to reports of drug-related liver injury leading to decompensated cirrhosis or liver failure.12,13 Patients with normal alkaline phosphatase (ALP) levels following UDCA therapy tend to have better long-term outcomes. 14 Therefore, current research considers changes in ALP levels as a primary outcome measure, alongside biochemical response rates and safety considerations.
Peroxisome proliferator-activated receptors (PPARs) are well-established nuclear hormone receptors crucial in regulating lipid metabolism, mitochondrial biogenesis, and energy homeostasis. 15 PAR agonists have demonstrated the ability to decrease bile acid toxicity, inflammation, and fibrosis by modulating nuclear receptor targets.16,17 Several randomized controlled trials (RCTs) have evaluated the efficacy and safety of PPAR agonists in patients who do not respond to UCDA compared to placebo. It was reported that more patients receiving PPAR agonists achieved composite biochemical response (ALP < 1.67 × upper limit of normal (ULN), ⩾15% ALP decrease from baseline, and total bilirubin ⩽ ULN).18,19 However, the impact of PPAR agonists on pruritus has shown varying results across different trials.17,20 In addition, the potential for PPARs to induce treatment-emergent adverse events (TEAE) remains inconclusively reported in various studies.17,19,20 The number of patients included in the current studies is limited. To address this limitation, meta-analysis can be used to amalgamate the results of current RCTs. Moreover, with multiple PPAR subtypes and both specific PPAR receptor type drugs and pan-PPAR inhibitors available, systematic reviews are necessary to assess the potential impact of different drugs on treatment effectiveness for PBC. Since the comprehensive and definitive evidence is limited, a systematic review and meta-analysis will be conducted to comprehensively evaluate the efficacy and safety of PPAR agonists in PBC.
Methods
We referred to the Cochrane Collaboration and the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) statement guidelines to develop this meta-analysis. 21 The protocol has been registered with PROSPERO (CRD42024545743). As this systematic review is based on published studies, no ethics approval is required. The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki.
Study objective
This study aims to assess the efficacy and safety of PPAR agonists in PBC. The primary outcomes are the reduction of ALP levels and biochemical response rates. The secondary outcomes are the rates of severe or serious AEs and relief of pruritus, which could be quantified by the Numeric Rating Scale (NRS), 5-D itch scale, or visual analog scale (VAS), depending on the assessment method employed in the studies.
Inclusion and exclusion criteria
The included studies were RCTs testing PPAR agonists in patients aged 18 years or older with PBC. For trials with multiple reports, only the most recent result was selected. Observational studies, editorials, commentaries, and review articles were excluded. Studies that reported at least one of the outcomes of interest in the original trials were included (eTable 1).
Data sources and search strategies
A comprehensive literature search was performed in PubMed, Web of Science, Embase, and Cochrane Central Register of Controlled Trials from the inception of each database to October 4, 2024. The detailed search strategies are described in eTable 2.
Study selection
Screening and selection were conducted by two investigators (W.L. and S.F.) independently. After the evaluation of the title and abstract of searched studies, full texts of retained articles were screened according to the inclusion and exclusion criteria to obtain the eligible research. Discrepancies were resolved by discussion with the third one (Y.Y.).
Data extraction
From each study, the following data were reviewed and extracted: name of study, first author and year of publication, study design and blinding, study phase, target population, number of patients, age distribution, sex distribution, treatment type and duration, dosage, background therapy with UDCA, ALP level change or the baseline and endpoint values of ALP level, number of patients achieved biochemical response, number of severe AE or/and serious AE, the pruritus score change. Two independent investigators (W.L. and S.F.) extracted data using a standardized form: study characteristics, study design, baseline characteristics of patients, intervention details, and outcomes. Discrepancies were resolved by discussion or by a third-author arbitration (Y.Y.). Accuracy was ensured via cross-verification against source documents and electronic double-entry validation.
Risk of bias, heterogeneity, and statistical inconsistency
The risk of bias was evaluated for assessing the risk of bias according to the Cochrane Collaboration’s tool. 22 Sources of heterogeneity were explored through subgroup and sensitivity analyses. We applied subgroup analysis to explore plausible explanations for heterogeneity, including (1) type of drugs; (2) treatment durations. Treatment duration more than 24 weeks was defined as long term. To assess the robustness of the pooled effects, each study was excluded in turn, and the consistency of the results was compared.
Statistical analysis
Mean difference (MD) of ALP level change and pruritus score change with their 95% confidence intervals (CIs) and risk ratio (RR) with 95% CI for biochemical response rate, rate of severe AE, and rate of serious AE were calculated in the intervention and control group from each study. The Q test was applied to determine whether methodological heterogeneity exists. Random effect models were applied in all analyses regardless of heterogeneity scores, as it has been shown to provide more robust estimates compared to fixed-effects models. 23 Fixed-effects models were performed as a sensitivity test when heterogeneity was low (p-value >0.10 or I2 < 50%).24,25 Two-sided p < 0.05 was considered significant for the difference between treatments.
Results
Study characteristics
Our systematic search identified a total of 1711 studies. After removing 239 duplicates, 1472 studies were eligible for title and abstract screening. Subsequently, 21 studies underwent full-text review, of which 14 RCTs finally met the eligibility criteria and were included in the systematic review and meta-analysis (eFigure 1). Three studies were excluded due to duplicate reporting,26–28 and two studies were excluded because sufficient data could not be extracted.16,29 The characteristics of the 14 included studies are summarized in Table 1.17–20,30–39 A total of 1137 patients were included across these studies, with women accounting for 92.2% of participants. Among the included studies, research on fenofibrate31,32,36 (PPAR α agonist) and seladelpar (PPAR δ agonist)18,30,34 was conducted in three studies each. Elafibranor17,19 (PPAR α and δ agonist) was studied in two trials, and saroglitazar 20 (PPAR α and γ agonist) was investigated in one RCT. The pan-PPAR agonist, bezafibrate,33,35,37–39 was studied in five RCTs. Ten studies have a treatment duration exceeding 24 weeks,17,18,30–33,35,37–39 with six defining biochemical response as their primary outcome.17,18,30–33 Other studies focused on changes in ALP levels. Nine studies reported the biochemical response rates,17–20,30–34 11 reported the change in ALP levels,17–20,30,33–38 six reported the severe AEs,17,19,32,34,38,39 and eight reported the serious AEs.17–20,30,33,34,37
Main characteristics of included randomized clinical trials.
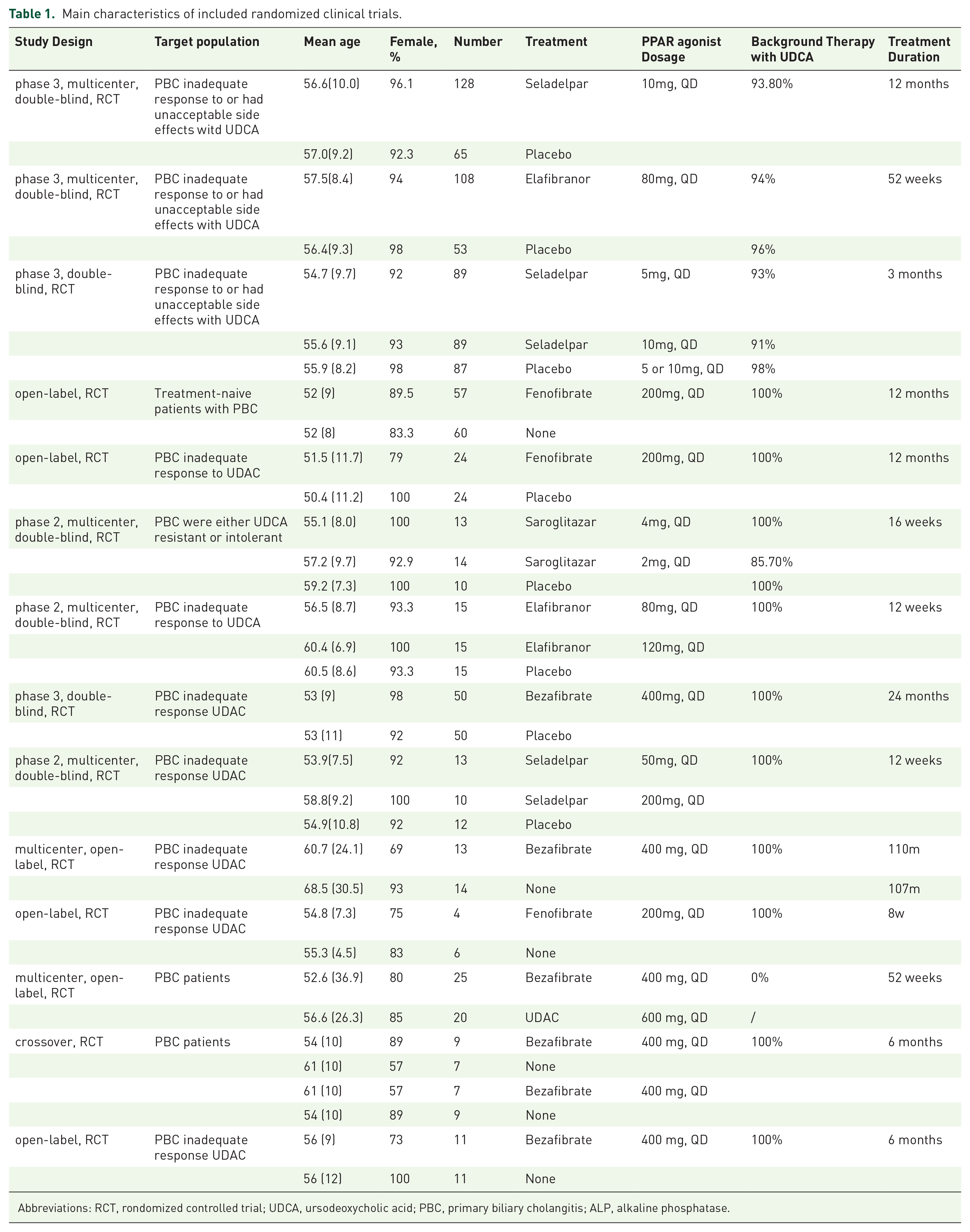
Abbreviations: RCT, rondomized controlled trial; UDCA, ursodeoxycholic acid; PBC, primary biliary cholangitis; ALP, alkaline phosphatase.
Primary efficacy outcomes
The effects of PPAR agonists on ALP levels are presented in Figure 1. Eleven studies reported the changes in ALP levels following treatment,17–20,30,33–38 and meta-analyses were performed. High heterogeneity was observed among the studies (I2 = 96%; p < 0.01). Random-effects model was used, and compared to control group, PPAR agonists significantly reduced ALP level by an MD of −155.87 U/L (95% CI, −208.30 to −103.44; random-effects). Data for the biochemical response rate in patients treated with PPAR agonists versus placebo were available from nine studies.17–20,30–34 Among the 860 patients, 534 received PPAR agonists, showing a significantly higher biochemical response rate (RR, 4.42, 95% CI, 2.37–8.26; random-effects; Figure 2). High heterogeneity was again observed among the studies (I2 = 79%; p < 0.01).

Meta-analysis of change in ALP level by PPAR agonists and placebo.

Meta-analysis of biochemical response rate by PPAR agonists and placebo.
Secondary efficacy outcomes
Different studies used various scales and criteria to evaluate the severity of pruritus and quality of life (eTable 3). At least one study utilized the NRS, Primary biliary Cirrhosis-40 (PBC-40) questionnaire, VAS, and 5-D itch scale. Three studies used the NRS to measure changes in pruritus among patients with a baseline NRS score of ⩾4.17,18,30 Specifically, treatment with seladelpar at a daily dosage of 10 mg significantly reduced the NRS score for pruritus, as reported in two RCTs.18,30 In one trial, the MD was −1.5 with a 95% CI of −2.5 to −0.5; in another, the MD was −1.59 with a p-value of 0.02. In contrast, seladelpar at a daily dosage of 5 mg (MD −0.46; p = 0.48) and elafibranor at a daily dosage of 80 mg (MD −0.78; 95% CI: −1.99 to 0.42) did not significantly alter the NRS score.17,18 Three studies employed the 5-D itch scale for assessment.17,30,34 Elafibranor at a daily dosage of 80 mg demonstrated significant improvements in pruritus compared to placebo (MD −3.0; 95% CI, −5.5 to −0.5). 17 However, seladelpar did not show efficacy in these assessments.30,34 In addition, bezafibrate at a daily dosage of 400 mg was also evaluated using the VAS, but the difference in changes from baseline to 24 months in itch intensity score between bezafibrate and placebo groups was not significant. 33
Safety outcomes
Regarding safety outcomes, six studies reported the severe AE,17,19,32,34,38,39 and eight studies reported the serious AE.17–20,30,33,34,37 Meta-analyses did not show any significant difference in the rate of severe AEs (RR, 1.05; 95% CI, 0.49–2.28; random-effects model) or serious AEs (RR, 1.02; 95% CI, 0.65–1.60; random-effects model; Figure 3). The 95% CI crossing 1 indicate no statistically significant difference in AE risk between PPAR agonists and control groups. The studies exhibited low heterogeneity (severe AE: I2 = 0%; p = 0.80; serious AE: I2 = 0%; p = 0.92). Hirschfield et al. 18 documented 56 treatment-related TEAEs, all of which were grade 1 or 2. The most frequently reported symptoms were pruritus and nausea. No serious treatment-related TEAEs were observed. Vuppalanchi et al. 20 reported 96 TEAEs in 31 patients, which were deemed unrelated to the study medication. However, serious TEAEs were reported in two patients in the saroglitazar group, manifesting as appendicitis, dyspnea, and peripheral edema. Schattenberg et al. 19 reported 37 patients with at least one TEAE, and eight of them experienced treatment-related TEAEs. One patient in the Elafibranor 120 mg group experienced a serious treatment-related TEAE. Notably, two patients treated with elafibranor experienced fatal AEs, 17 although neither was deemed related to treatment.

Meta-analysis of adverse event rates. (a) Severe AE. (b)Serious AE.
Risk of bias
We assessed the risk of bias in eligible RCTs using the Cochrane Collaboration’s tool (eFigure 2). All studies demonstrated a low risk of attrition bias and reporting bias. While most studies showed a low risk of selection bias related to random sequence generation, some did not clearly describe their randomization methods, resulting in unclear selection bias.17,20,36–39 Four studies displayed an unclear risk of selection bias related to allocation concealment.36–39 Seven RCTs had a high risk of performance bias and detection bias due to the absence of double-blindness.31,32,35–39 Furthermore, six studies were considered to have an unclear risk of other biases associated with sponsorship.17–20,30,34 One crossover RCT was classified as high risk of other biases due to the potential residual drug effect. 38
Subgroup analyses and sensitivity analyses
Overall, the funnel plot demonstrated an asymmetrical distribution of the published studies (eFigure 3). Subgroup analyses and sensitivity analyses were conducted to investigate the source of heterogeneity. When sequentially excluding each trial, the results of meta-analyses supported the finding that PPAR agonists can significantly reduce ALP levels (eFigure 4). Subgroup analyses based on drug type and treatment duration yielded consistent conclusions (eFigures 5 and 6). For patients treated with bezafibrate, PPAR agonists significantly reduced the ALP level by a MD of −217.14 U/L (95% CI, −346.16 to −88.13; random-effects). Similarly, for patients treated with seladelpar, a significant reduction in ALP level was observed with an MD of −124.98 U/L (95% CI, −152.71 to −97.25; random-effects). When a treatment duration exceeding 24 weeks was defined as long-term, long-term treatment with PPAR agonists resulted in a significant reduction in ALP levels by a MD of −183.40 U/L (95% CI, −276.69 to −90.11; random-effects model), along with an improved biochemical response rate (RR 3.58, 95% CI, 1.51–8.51; random-effects model). Meanwhile, short-term treatment with PPAR agonists also significantly reduced the ALP level by a MD of −130.45 U/L (95% CI, −157.50 to −103.40; random-effects), and an enhanced biochemical response rate (RR, 6.13, 95% CI, 3.34–11.24; random-effects).
Discussion
The primary outcome of this meta-analysis is that in patients with PBC, PPAR agonists significantly reduced the ALP levels and improved the biochemical response rates, irrespective of drug type and treatment duration. Notably, the rates of serious AEs and severe AEs are comparable to placebo. Recent network meta-analyses have established the biochemical benefits of PPAR agonists in UDCA-refractory PBC.40,41 However, they have not systematically examined patient-reported symptom relief, agent-specific safety, or pharmacologic consistency. Our systematic review expands this evidence base in three ways: First, we incorporate patient-centered outcomes using four validated pruritus scales (e.g., NRS, 5-D), demonstrating significant itch relief with seladelpar and elafibranor. Second, we quantify severe and serious AEs across agents, showing comparable safety to placebo. Third, our drug-specific and duration-stratified analyses confirm robust and consistent efficacy. These contributions fill critical gaps in prior analyses and provide integrated data for more nuanced, evidence-based therapeutic decision-making in PBC.
PPARs play a crucial role in regulating inflammation, carcinogenesis, and metabolism. 42 PPAR-α, γ, and δ all contribute to reducing inflammation. In addition, PPAR-α is involved in regulating lipid metabolism, reducing liver fibrosis, and controlling bile acid metabolism; PPAR-γ can increase steatosis and has antifibrotic activity; PPAR-δ can decrease steatosis and protect against carcinogenesis.43,44 Overall, PPAR agonists show promise as molecular targets in PBC.
We also examined whether PPAR agonists target different isoforms. From the subgroup analysis of meta-results, it was found that both elafibranor and seladelpar significantly reduced ALP levels. Similarly, the results of a single RCT for fenofibrate and saroglitazar also showed significant reductions in ALP levels. However, there was one RCT using bezafibrate where the authors reported a reduction in serum ALP (−35%, p = 0.03 vs placebo), but our calculation resulted in an MD of −94.5 (95% CI, −227.10 to 38.10). 16 While bezafibrate was able to reduce ALP compared to placebo, there was no statistically significant difference which may be due to the shorter treatment period (21 days) compared to all other studies or because this study did not limit inclusion based on inadequate response or unacceptable side effects with UDCA as baseline therapy which could have masked the effect of bezafibrate. In terms of biochemical response rate, both Elafibranor and Seladelpar’s subgroup analysis results, as well as the single RCT result of bezafibrate, showed significantly higher response rates in the experimental group compared to control.
Pruritus is a significant issue for patients with PBC and is a secondary outcome of interest in this study. Early pruritus may portend a more severe disease course and a less favorable response to treatment, highlighting symptom improvement as a clinically relevant surrogate marker for disease progression. 45 Due to variations in the study population (all patients or those with moderate to severe pruritus at baseline) and the use of diverse scales for assessing pruritus, we did not conduct a meta-analysis on this aspect but instead listed the results in eTable 3. The primary outcome of the RCT designed by de Vries et al. 16 was the improvement of pruritus after short-term treatment (21 days) with PPAR agonists (⩾50% reduction of pruritus). The results showed that bezafibrate led to a 45% improvement (41% in PSC, 55% in PBC), compared to 11% with placebo as the primary outcome (p = 0.003). Furthermore, all studies targeting patients with moderate to severe baseline pruritus demonstrated the ability of PPAR agonists to alleviate itching.
As UDCA is the first-line treatment for PBC, studies have focused only on PBC patients who had an inadequate response or experienced unacceptable side effects from UDCA. Currently, meta-analysis results indicate that PPAR agonist therapy for PBC is effective and safe, with potential for relieving pruritus. However, one limitation is that individual RCT has small sample sizes; future accumulation of data from larger samples will support its effectiveness and safety. This may lead to considering direct use of PPAR agonists for PBC patients and obtaining data to support whether it can be considered as a first-line treatment option for them.
Our meta-analysis has several limitations that should be noted. First, the results are based on study-level data rather than individual data, although we synthesized high-quality evidence from double-blind RCTs. Second, there are limitations in terms of study numbers, PBC subtype, treatment duration, and outcomes. Our meta-analysis included only 14 studies with a total of 1137 participants, which constrains the statistical power for subgroup analyses and rare safety outcomes. More critically, the analysis predominantly reflects classic PBC phenotypes. The majority of included trials systematically excluded patients with variant disease forms such as PBC-autoimmune hepatitis overlap (PBC-AIH) and ductopenic variants. Of the 14 studies, 7 explicitly excluded patients with autoimmune hepatitis or PBC-AIH, while 4 others broadly excluded those with “other liver diseases”—effectively omitting these clinically distinct subgroups. This exclusion is methodologically significant given that PBC-AIH diagnosis relies on mandatory histological assessment and validated tools like the simplified AIH scoring system. 46 Consequently, our findings cannot be generalized to overlap syndromes or atypical variants, which represent therapeutic challenges with disproportionately poor outcomes. Future trials should prioritize the inclusion of these high-risk subgroups through deliberate enrollment strategies and standardized histological criteria to better define PPAR agonists’ role across the PBC spectrum. More data from PBC patients is needed to support the efficacy and safety of PPAR agonist therapy. Additionally, the treatment duration varied greatly among the included studies, ranging from as short as 21 days to as long as 2 years. More long-term efficacy data are required for future analysis. Furthermore, different itch severity assessment scales were used in the studies. Finally, high heterogeneity was observed among the studies when evaluating the change in ALP. Therefore, subgroup analyses and sensitivity analyses were conducted. Overall, the findings should be interpreted with caution due to these limitations, and further research addressing these issues would provide more robust evidence for clinical practice and decision-making processes related to PPAR agonist therapy for PBC patients.
Conclusion
In patients with PBC, PPAR agonists significantly reduced the ALP levels and improved the biochemical response rates, irrespective of drug type and treatment duration. PPAR agonists appear safe and well-tolerated by patients with PBC.
Supplemental Material
sj-docx-1-tag-10.1177_17562848251370111 – Supplemental material for Efficacy and safety of PPAR agonists in primary biliary cholangitis: a systematic review and meta-analysis
Supplemental material, sj-docx-1-tag-10.1177_17562848251370111 for Efficacy and safety of PPAR agonists in primary biliary cholangitis: a systematic review and meta-analysis by Wanying Liao, Siyang Fu, Aiming Yang and Yingyun Yang in Therapeutic Advances in Gastroenterology
Supplemental Material
sj-docx-8-tag-10.1177_17562848251370111 – Supplemental material for Efficacy and safety of PPAR agonists in primary biliary cholangitis: a systematic review and meta-analysis
Supplemental material, sj-docx-8-tag-10.1177_17562848251370111 for Efficacy and safety of PPAR agonists in primary biliary cholangitis: a systematic review and meta-analysis by Wanying Liao, Siyang Fu, Aiming Yang and Yingyun Yang in Therapeutic Advances in Gastroenterology
Supplemental Material
sj-jpg-2-tag-10.1177_17562848251370111 – Supplemental material for Efficacy and safety of PPAR agonists in primary biliary cholangitis: a systematic review and meta-analysis
Supplemental material, sj-jpg-2-tag-10.1177_17562848251370111 for Efficacy and safety of PPAR agonists in primary biliary cholangitis: a systematic review and meta-analysis by Wanying Liao, Siyang Fu, Aiming Yang and Yingyun Yang in Therapeutic Advances in Gastroenterology
Supplemental Material
sj-jpg-3-tag-10.1177_17562848251370111 – Supplemental material for Efficacy and safety of PPAR agonists in primary biliary cholangitis: a systematic review and meta-analysis
Supplemental material, sj-jpg-3-tag-10.1177_17562848251370111 for Efficacy and safety of PPAR agonists in primary biliary cholangitis: a systematic review and meta-analysis by Wanying Liao, Siyang Fu, Aiming Yang and Yingyun Yang in Therapeutic Advances in Gastroenterology
Supplemental Material
sj-jpg-4-tag-10.1177_17562848251370111 – Supplemental material for Efficacy and safety of PPAR agonists in primary biliary cholangitis: a systematic review and meta-analysis
Supplemental material, sj-jpg-4-tag-10.1177_17562848251370111 for Efficacy and safety of PPAR agonists in primary biliary cholangitis: a systematic review and meta-analysis by Wanying Liao, Siyang Fu, Aiming Yang and Yingyun Yang in Therapeutic Advances in Gastroenterology
Supplemental Material
sj-jpg-5-tag-10.1177_17562848251370111 – Supplemental material for Efficacy and safety of PPAR agonists in primary biliary cholangitis: a systematic review and meta-analysis
Supplemental material, sj-jpg-5-tag-10.1177_17562848251370111 for Efficacy and safety of PPAR agonists in primary biliary cholangitis: a systematic review and meta-analysis by Wanying Liao, Siyang Fu, Aiming Yang and Yingyun Yang in Therapeutic Advances in Gastroenterology
Supplemental Material
sj-jpg-6-tag-10.1177_17562848251370111 – Supplemental material for Efficacy and safety of PPAR agonists in primary biliary cholangitis: a systematic review and meta-analysis
Supplemental material, sj-jpg-6-tag-10.1177_17562848251370111 for Efficacy and safety of PPAR agonists in primary biliary cholangitis: a systematic review and meta-analysis by Wanying Liao, Siyang Fu, Aiming Yang and Yingyun Yang in Therapeutic Advances in Gastroenterology
Supplemental Material
sj-jpg-7-tag-10.1177_17562848251370111 – Supplemental material for Efficacy and safety of PPAR agonists in primary biliary cholangitis: a systematic review and meta-analysis
Supplemental material, sj-jpg-7-tag-10.1177_17562848251370111 for Efficacy and safety of PPAR agonists in primary biliary cholangitis: a systematic review and meta-analysis by Wanying Liao, Siyang Fu, Aiming Yang and Yingyun Yang in Therapeutic Advances in Gastroenterology
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.