
Abstract
Hereditary hemorrhagic telangiectasis (HHT) and juvenile polyposis syndrome (JPS) are both relatively rare hereditary disorders. It has been reported that patients with SMAD4 mutations may suffer from both HHT and JPS, defined as JPS/HHT. To improve the understanding and diagnosis of these diseases, we herein report a case of a 17-year-old male with abdominal pain and hematochezia. Low-tension computed tomography (CT) of the small intestine showed intussusception. Combined with the patient’s medical history of nasal bleeding and pulmonary arteriovenous fistula (pAVF) embolism, a final diagnosis of JPS/HHT was reached, according to the Curaçao Diagnostic Criteria. The possibility of JPS/HHT should be considered in patients with epistaxis and intussusception.
Keywords
Introduction
Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant disorder that causes anomalous angiogenesis, characterized by telangiectasia and arteriovenous malformations (AVMs). The most common clinical manifestation is spontaneous and recurrent rhinorrhagia (epistaxis) beginning at an average age of 12 years. Telangiectasia, usually found in the lips, tongue, face, fingers, buccal and gastrointestinal (GI) mucosa, and nasal mucosa, usually appears later than epistaxis. 1 Juvenile polyposis syndrome (JPS) is an autosomal dominant genetic disease characterized by the development of histopathological juvenile polyps throughout the GI tract, which can present as abdominal pain, diarrhea, GI bleeding, anemia, prolapsed rectal polyp, intestinal obstruction, or intussusception. 2 It has been reported that patients with SMAD4 mutations may suffer from both HHT and JPS, defined as JPS/HHT.
Here, we present a case of 17-year-old male with abdominal pain and hematochezia due to JPS/HHT.
Methods
The present case report is designed following the CAse REport (CARE) guidelines. 3 The CARE checklist of information to be included when writing a case report is added as a Supplemental File. We obtained written informed consent from the patient to publish the data, and we de-identified the data as far as possible. An ethics approval was not necessary for this case report.
Case report
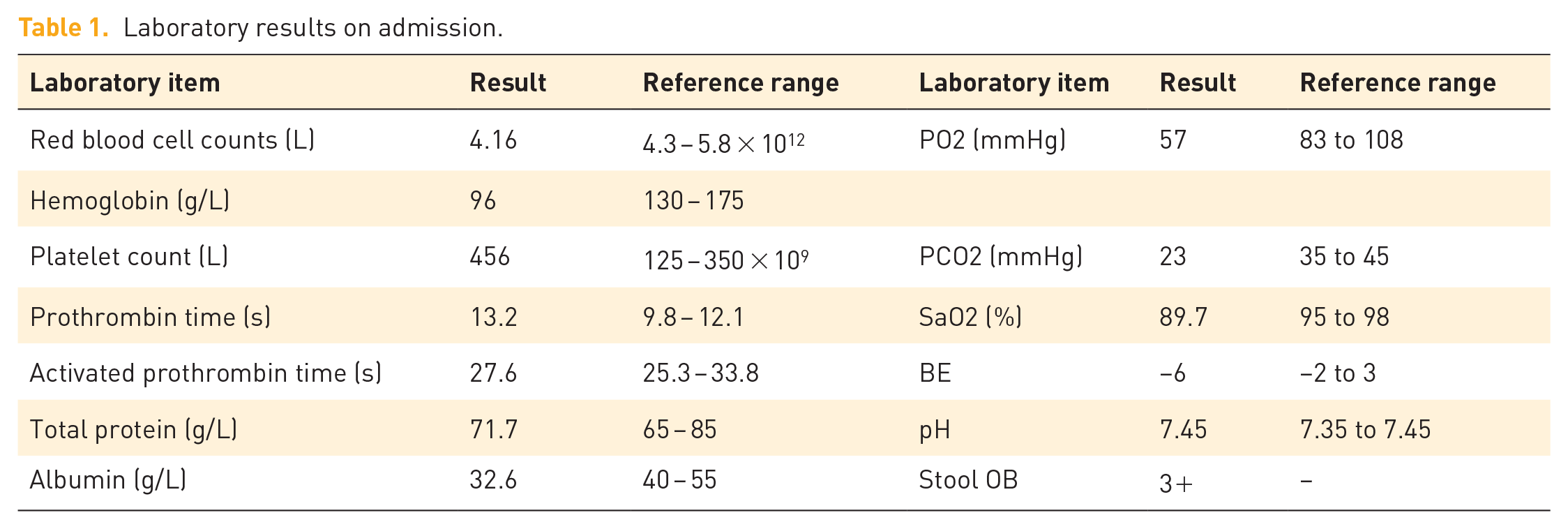
A 17-year-old male was admitted to our hospital due to changes in stool characteristics over a period of 1 month; the patient reported passing shapeless, and bloody stools 2–3 times a day alongside intermittent abdominal colic and abdominal distension. Local colonoscopy revealed multiple colonic polyps, that required further treatment in our hospital. The patient had a history of pulmonary arteriovenous fistula (pAVF), underwent intestinal polypectomy at 2 years of age, and underwent pAVF embolization 5 years ago. The patient reported that he was prone to having his finger, toes, and lips turn blue, which was obvious after substantial activity, and was prone to spontaneous nasal bleeding from childhood. However, the patient did not pay attention to these symptoms and did not seek further treatment. The patient was the only child in his family and his family had no similar diseases. Physical examination (Figure 1) showed clubbed nails on all four limbs, pectus carinatum with lordosis, right lower abdominal tenderness, and no rebound pain. Other vital signs were normal. Laboratory results on admission showed a decreased albumin level and mild anemia (Table 1), and urine and tumor markers showed no obvious abnormalities. Gastroscopy showed telangiectasia of the duodenum and fundus of the stomach [Figure 2(a) and (b)], and multiple colon polyps with a long, stalk-like and lobulated appearance [Figure 2(c) and (d)]. Pulmonary artery computed tomography angiography (CTA) showed multiple pulmonary AVMs in both lungs (Figure 3(a) and (b)). Low-tension CT of the small intestine (Figure 3(c) and (d)) showed multiple polyps and right abdominal intussusception, and a right portal vein right hepatic vein fistula was considered. Brain magnetic resonance imaging and magnetic resonance angiography (MRI+MRA) did not find cerebral AVMs.

(a and b) Clubbed nails on all four limbs and (c) Pectus carinatum.
Laboratory results on admission.

Endoscopy showed (a and b) telangiectasias in the duodenum and stomach and (c and d) multiple juvenile polyps in the intestine.

CTA of both lungs showed (a and b) obvious thickening, an arteriovenous anastomosis, forming a malformed vascular mass shadow, and “I-shaped” embolism nail. Abdominal enhanced CT showed (c and d) intussusception with significant thickening and enhancement of the local intestinal wall, suggesting multiple polyps.
Based on the above findings, the patient met the diagnostic criteria for JPS/HHT.4,5 We informed all treatment options with the patient, who favored the surgical therapy, and we obtained patient’s consent for the treatment. One month later, the patient underwent a right hemicolectomy and ileocolon-transverse colon anastomosis. The patient was discharged after treatment, with improved nutritional support. Six months post-surgery, the patient’s abdominal symptoms improved without abdominal pain, abdominal distension, bloody stools and other symptoms, and his body weight increased by 5 kg.
Literature review and discussion
HHT can be diagnosed by genetic testing or clinically based on the Curaçao Diagnostic Criteria: A definite diagnosis is made in the presence of at least three of the following manifestations: spontaneous and recurrent rhinorrhagia (epistaxis); telangiectasias in multiple areas (especially at characteristic sites: lips, oral cavity, fingers, or nose); visceral lesions with or without hemorrhage (pulmonary, hepatic, cerebral, or spinal AVMs), and family history (an immediate family member with HHT), and consider a probable or suspected diagnosis when two criteria are present. 5 The patient met the first three diagnostic criteria of Curaçao, which corresponded to a clear diagnosis of HHT. For patients with a definite clinical diagnosis, genetic testing is not required to confirm the diagnosis. 6
If the patient meets any of the following three clinical diagnostic criteria, 4 JPS can be diagnosed: (i) ⩾5 juvenile polyps in the large intestine; (ii) multiple juvenile polyps throughout the whole digestive tract; or (iii) a family history of JPS and regardless of the number of juvenile polyps.
Currently, there are five known HHT genotypes, of which only three are related to specific genes. ENG mutations are associated with HHT1 type, which often manifests as an arteriovenous fistula in the lung and brain. The ACVRL1 gene mutation is associated with HHT2, which is usually characterized by AVM of the liver and spinal cord, as well as nasal bleeding and pulmonary hypertension.7,8 HHT3 and HHT4 are not associated with specific genes. Although JPS and HHT were first reported as overlapping syndromes in 1980, 9 it was only in 2004 that Gallione et al. found that JPS/HHT onset was related to SMAD4 mutations. 10
Our case is particularly novel due to the lack of an identifiable SMAD4 mutation; there are relatively few reports of such patients in the literature. This case sheds new light on the diversity of genetic backgrounds underlying JPS/HHT. Furthermore, the lack of any family history for any of the symptoms associated with this disease is unusual considering the typical inheritance pattern.
This case is also unique because there are few case reports of JPS/HHT in Asian patients. A literature search revealed that studies of JPS/HHT are reported less, mostly from Western countries, while there are rare reports from East Asian countries.
Hashimoto et al. 11 reported a case of multiple polyps detected on colonoscopy due to suspected Crohn’s disease. No mutations were found in the familial adenomatous polyposis or Peutz–Jeghers syndrome-related gene tests. Many years later, due to nasal bleeding and telangiectasia of the maxillary and gastric capillaries, SMAD4 gene testing showed a missense mutation from arginine to cysteine at codon 361, and JPS/HHT was diagnosed.
Kang et al. 12 reported a case in South Korea of a hereditary hemorrhagic patient with blood capillary dilation and juvenile polyposis. The patient’s frequent nose bleeding occurred at the age of 5 and JPS symptoms began at the age of 7 years; 30–50 colon polyps were observed. Histological examination confirmed juvenile polyp specimens after resection. Physical examination revealed telangiectasia of the lip and tongue and bulbous protrusions of the extremities. Capsule endoscopy showed 10 small polyps and telangiectasia in the jejunum. Sanger sequencing then revealed a new SMAD4 mutation.
Liu et al. 13 reported a case of JPS accompanied by HHT syndrome. The patient underwent digestive endoscopic forceps removal for “multiple polyps of the colon” 3 years previously, and occasionally had black stools after surgery. Digestive system ultrasound suggested secondary chronic loose intussusception. Abdominal ultrasound showed a small nodular hyperechoic appearance in the right lobe of the liver, suggesting hemangioma. The final total exon gene assay suggested heterozygous variation in SMAD4.
As a result of arteriovenous fistulas, patients with HHT are more likely to present with disease features such as brain abscesses, migraines, ischemic/embolic strokes, cardiac failure, and widespread bleeding complications. 14 Treatment for pulmonary AVMs is required when the size of supply vessels are ⩾3 mm in size or if the treatment is technically feasible. 1 In this case, the patient was diagnosed with pAVF and underwent embolization. The symptoms improved after treatment, but the clubbing caused by long-term hypoxia was irreversible. Ospina et al. 15 reported amelioration of respiratory symptoms due to diffuse pulmonary AVM treated with bevacizumab, reduction in nasal bleeding, and no new AVM formation on chest CT during follow-up. This provides a new treatment strategy for diffuse pulmonary AVM. Current recommendations for GI management of JPS include colonoscopy every 2–3 years and upper endoscopy every 1–3 years, starting with symptoms or in adolescence if asymptomatic.16,17
Experience and inadequacies
A review of the history showed that the patient had been diagnosed with intussusception for more than 10 years from the onset of the disease to the presentation of intussusception. This experience is worth summarizing as JPS–HHT is a rare disease in clinical practice, and clinicians have insufficient understanding of the JPS/HHT syndrome. The patient first presented to a doctor with purple lips and the clinician first considered common diseases of the heart and lungs, and performed the relevant examinations. Finally, a pAVF was diagnosed and embolization was performed. This finding is consistent with conventional clinical diagnostic thinking. According to a single-center study in the United States, 18 nosebleeds and pulmonary AVMs are the most frequent clinical manifestations of HHT/JPS, with about 77% of patients having prior nosebleed and 60% of patients with visceral AVM. The preferred sites of visceral AVM were the lower respiratory system (45%), liver (27%), and GI tract (10%). Combined with the patient’s history of repeated epistaxis, a diagnosis of HHT was not difficult. The patient was also not consulted carefully enough, and important past medical history that he had undergone an intestinal polypectomy when he was 2 years old was overlooked. Nowadays, the division of departments is becoming increasingly common, which often results in failures in the collection of history from other related departments and missed associations with two or more diseases with a diagnosis. In this case, intestinal polyps were present at an early stage but were overlooked as unimportant. The limitation of this study was that the patient did not undergo genetic testing to confirm the diagnosis due to economic reasons.
Conclusion
JPS/HHT remains a rare disease in clinical, especially in the East Asian countries. Reports of recurring unexplained nasal bleeding, oral cyanosis, and clubbing symptoms in patients should alert to the potential possibility of HHT. When patients have GI telangiectasia and juvenile polyposis, organs such as the liver, lungs, and brain should be evaluated to determine the possibility of JPS/HHT.
Supplemental Material
sj-docx-1-tag-10.1177_17562848221142913 – Supplemental material for Hereditary hemorrhagic telangiectasis with juvenile polyposis syndrome: a case report
Supplemental material, sj-docx-1-tag-10.1177_17562848221142913 for Hereditary hemorrhagic telangiectasis with juvenile polyposis syndrome: a case report by Meng-Yu Tao, Kai-Yi Wang, Xin Li, Chen Yu, Qin-Si Wan, Xu Shu, You-Xiang Chen and Wang-Di Liao in Therapeutic Advances in Gastroenterology
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.