
Abstract
Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant vascular disease. ENG and ACVRL1 gene variants account for up to 96% of all cases, while the remaining cases are caused by SMAD4 or GDF2 variants, or by currently undiscovered mutations in coding or non-coding regions. Here, we report a 47-year-old man who presented with duodenal bulb bleeding and chronic anemia. Physical examination also revealed bleeding from the skin and gingiva. His parents were cousins and one brother and one sister died in infancy from anemia and bleeding. Head computed tomography angiography (CTA) revealed a complete fetal posterior cerebral artery located in the left side, and pulmonary CTA showed pulmonary arterial hypertension. The patient was diagnosed with HHT. Peripheral blood was collected for whole-exome sequencing. Sequencing revealed a mutation in the GDF2 gene, which encodes bone morphogenetic protein-9 (BMP-9). The detected variant, c.352A > T(p.Ile118Phe), was predicted to be a neutral polymorphism; however, the patient’s plasma BMP-9 levels were greatly reduced; we predicted that this might be caused by the GDF2 variant and might be involved in the HHT pathogenesis. Further research in cell lines and animal models is needed to verify the correlation between this GDF2 variant and the pathogenesis of HHT.
Keywords
Introduction
Hereditary hemorrhagic telangiectasia (HHT) is a rare autosomal dominant condition, also known as Osler–Weber–Rendu disease, with an estimated global prevalence of 1:5000–1:8000 individuals.1,2 HHT is frequently misdiagnosed by clinicians due to its lack of specific clinical symptoms. 3 Symptoms associated with this disorder include mucocutaneous telangiectasias, epistaxis, and arteriovenous malformations (AVMs) in the lungs, liver, brain, and spine.4–6 Family history and gene variant testing are crucial for the diagnosis of HHT. Variants in three key genes are currently known to cause HHT: ENG, encoding endoglin (HHT1), ACVRL1, encoding activin A receptor type-like kinase 1 (HHT2), and SMAD4 (juvenile polyposis-HHT).7,8 GDF2, which encodes growth differentiation factor 2/bone morphogenetic protein-9 (BMP-9; HHT5), is also associated with an HHT-like phenotype. 9 Here we report on a patient with HHT who carried the missense variant c.352A > T(p.Ile118Phe) in the GDF2 gene.
Case Report
The reporting of this study conforms to the CARE guidelines. 10 The study was reviewed and approved by the Ethics Committee of the First Affiliated Hospital of the Army Medical University (Approval No. (B)KY202283). Written informed consent was obtained from the patient for publication in this study.
A 47-year-old Chinese male patient attended the emergency department of our hospital with a 10-day history of dizziness, fatigue, and hematemesis. He also reported a history of anemia for more than 40 years and a long history of spontaneous and recurrent epistaxis. Blood tests showed a white blood cell count of 6.51 × 109/L, red cell count 1.67 × 1012/L, hemoglobin 32 g/L, mean corpuscular volume 74.30 fL, mean corpuscular hemoglobin 19.20 pg, and platelets 81 × 109/L. The patient was given an infusion of 8 U of red blood cells to correct his anemia. Gastroscopy indicated gastric mucosal hemorrhage (Figure 1a). Physical examination revealed bleeding from the gums and mouth (Figure 1b), scattered telangiectasias on the arm (Figure 1c), and a subcutaneous hematoma on the left knee (Figure 1d). Head computed tomography angiography (CTA) revealed a complete fetal posterior cerebral artery (cfPCA) located on the left side (Figure 1e). Pulmonary CTA showed pulmonary arterial hypertension (PAH).

Clinical presentation of the patient. (a) Gastroscopy showed gastric mucosa hemorrhage. Physical examination revealed (b) bleeding of the gums and mouth, (c) subcutaneous hemorrhagic spots on the arms, (d) subcutaneous hematoma on the left knee and (e) Head computed tomography angiography showed a complete fetal posterior cerebral artery located on the left side.
The family had a history of consanguineous marriage. His grandmothers on his mother's and father's sides were sisters. His parents and grandparents had no HHT-related symptoms. He had three sisters and one brother; one brother and one sister had similar HHT-related symptoms and died because of bleeding at a young age (Figure 2a).
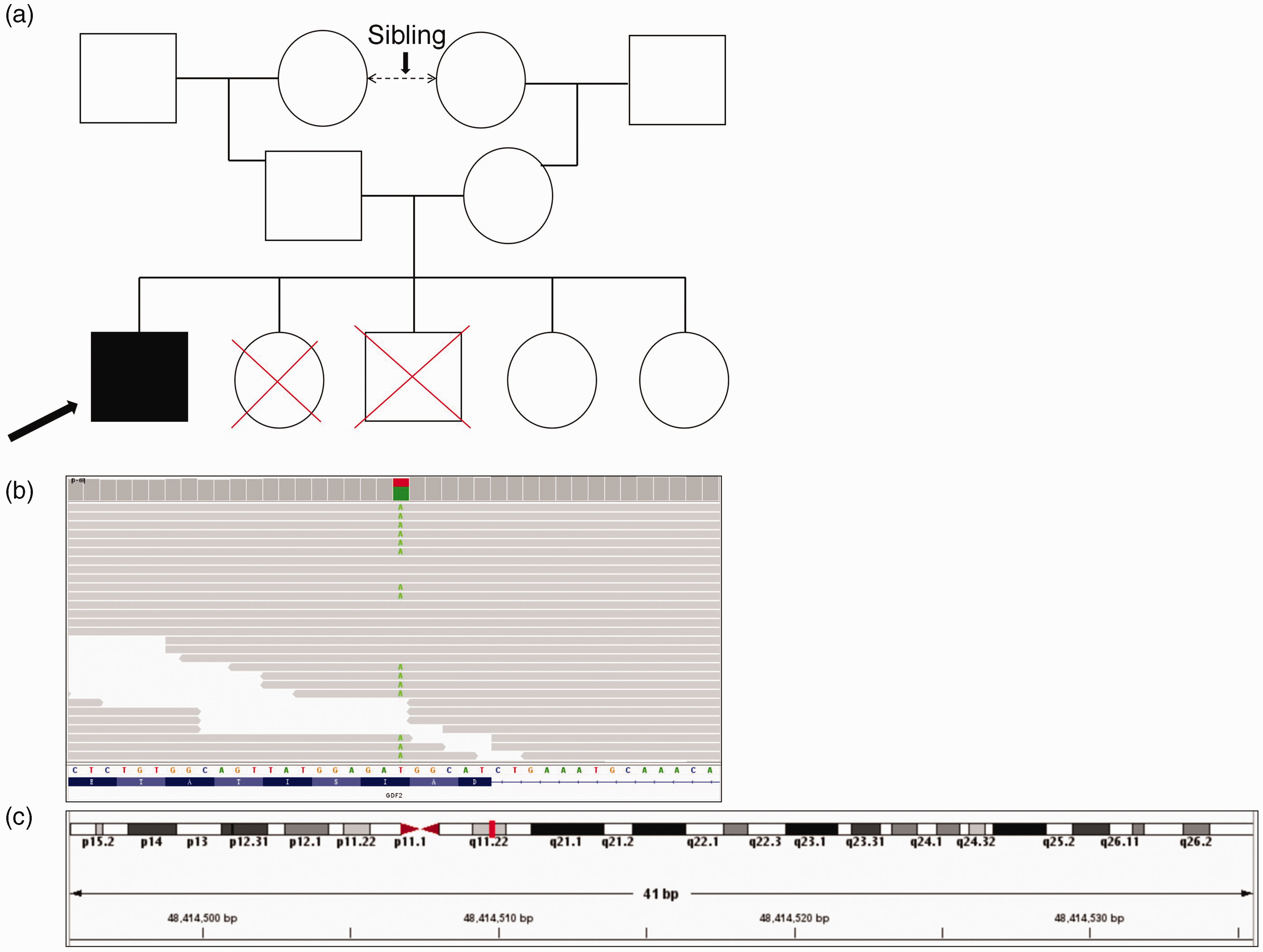
Family tree and whole-exome sequencing data of the GDF2 gene in the patient. (a) The proband’s (arrowhead) family pedigree revealed first‐cousin consanguinity. Circle: female, square: male, red cross: bled to death. (b) Analysis of whole-exome sequencing data by Integrative Genomics Viewer confirmed a heterozygous GDF2 mutation and (c) Homozygosity mapping revealed that the mutant GDF2 gene (red blocks) was located on chromosome 10.
We collected peripheral blood from the patient for whole-exome sequencing (WES), which identified 243,479 sequence variants, including 26,786 exonic and splicing variants (Table 1). Diagnostic exome sequencing and bioinformatics analyses revealed the GDF2 variant chr10:48414516 NM_016204.4:c.352A > T(p.Ile118Phe) (Figure 2b). This variant was confirmed to be heterozygous in this patient using the Integrative Genomics Viewer. This missense mutation in GDF2 was located within a 41-bp region of chromosome 10 (Figure 2c). The gnomAD database recorded this GDF2 variant in 310 heterozygous individuals and five homozygous individuals, with a frequency of 0.0111%, and predicted that it was a neutral polymorphism. Notably however, plasma BMP-9 levels were markedly reduced in the current patient compared with healthy donors (61.85 ± 7.56 pg/mL versus 211.47 ± 13.71 pg/mL). The missense mutation (p.Ile118Phe) in the BMP-9 protein was located within its conserved prodomain sequence (Figure 2c), and may thus affect the expression of the mature protein.
Summary of exon-sequencing results.

MAF, minor allele frequency.
In addition, exome sequencing and bioinformatics analyses revealed two variants in the CUBN gene (encoding cubilin): chr10:16949651 NM_001081.3:c.7561C; > G(p.Pro2521Ala) and chr10:16957924 NM_001081.3:c.7106G>T(p.Gly2369Val) (Figure 3a), which were confirmed as heterozygous by Integrative Genomics Viewer analysis. These two missense mutations in CUBN were also located on chromosome 10 (Figure 3b). However, CUBN mutations have not been reported to be associated with HHT.

Whole-exome sequencing analysis of the CUBN gene in the patient. (a) Analysis of whole-exome sequencing data by Integrative Genomics Viewer confirmed two heterozygous CUBN mutations and (b) Homozygosity mapping revealed that the mutant CUBN gene (red blocks) was located on chromosome 10.
The patient was discharged from hospital following examinations and blood transfusion treatment.
Discussion
HHT is an inherited blood vessel disease, and symptoms develop gradually with age. 11 The Curacao criteria are commonly used in the clinical diagnosis of HHT 3 : (i) recurrent and spontaneous epistaxis; (ii) multiple mucocutaneous telangiectasias (nose, fingers, oropharynx, and lips); (iii) brain, spinal, pulmonary, and hepatic AVMs and gastrointestinal telangiectasia; and (iv) a family history of HHT. Establishment of a diagnosis requires three criteria to be met, while two criteria indicates a possible diagnosis of HHT, but less than two criteria means a diagnosis is unlikely.
Advances in experimental technology have increased the importance of gene sequencing for diagnosing diseases. In 1980, Cox et al. identified a novel case of an AVM associated with a genetic mutation and suggested that this represented a new hereditary syndrome. 12 Most HHT patients present with a heterozygous pathogenic gene variant. 13 Previous studies found that ENG (HHT1) variants may be responsible for cerebral and pulmonary AVMs. 14 ENG variants have been reported in more than 500 patients with HHT, including insertions, deletions, copy number variations, and nonsense variants. 15 The ACVRL1 (12q13.13) mutation is also an important variant in HHT patients. 16 Many patients with ENG and ACVRL1 mutations can present with the same symptoms, and over 750 pathogenic ENG and ACVRL1 variants have been described to date. 17 If HHT is highly suspected based on clinical symptoms but the individual tests negative for ENG and ACVRL1 variants, SMAD4 may also be examined.18,19 At least one pathological variant in ENG, ACVRL1, or SMAD4 can be detected by sequence analysis in over 85% to 90% of HHT patients.20–22 Previous studies indicated that GDF2 analysis may also be considered in HHT patients without pathologic variants in the above three genes, particularly in patients with subcutaneous hemorrhages that are not limited to the mouth, hands, and face.23,24 GDF2 (located at 10q11.22) has been reported to play important roles in the development of the vascular architecture.9,25 Balachandar et al. 26 reported a case in which WES identified a novel heterozygous GDF2 variant in three patients from a single HHT family, all of whom screened negative for SMAD4, ACVRL1, and ENG variants, and all of whom had typical HHT symptoms and severe pulmonary AVMs. Gallego et al. 27 identified a homozygous missense GDF2 variant in a pediatric patient diagnosed with PAH associated HHT. Hodgson et al. 28 reported one homozygous nonsense GDF2 gene variant in two pediatric patients, one with pulmonary AVMs and the other with PAH, both with facial telangiectasias and low levels of circulating BMP-9. Wang et al. 29 reported a 5-year-old boy who was diagnosed with severe PAH in whom genetic screening revealed a novel homozygous nonsense mutation in the GDF2 gene, which was also present in his parents.
In the current case, we identified a low-frequency GDF2 missense mutation in a patient with HHT. The variant was predicted to be a neutral polymorphism according to the gnomAD database; however, the patient’s BMP-9 plasma levels were lower than normal. We considered that the GDF2 mutation may be the cause of the decrease in BMP-9 levels, and may be involved in the pathogenesis of HHT. Moreover, we also detected two low-frequency missense CUBN mutations, which were predicted to be benign according to the American College of Medical Genetics and Genomics guidelines. Previous studies described causative CUBN variants in Imerslund-Grasbeck Syndrome, 30 which has been implicated as a hereditary cause of variable proteinuria and megaloblastic anemia. 31 Currently however, CUBN variants have not been associated with HHT. The present patient had anemia but not megaloblastic anemia. There is currently insufficient evidence to prove a correlation between CUBN variants and HHT.
This case report had two limitations. First, we could only speculate that the detected GDF2 variant played a role in the pathogenesis of HHT. We subsequently aim to verify the correlation between the GDF2 variant and HHT pathogenesis in cell lines and mouse models. Second, HHT is regarded as an autosomal dominant disorder; however the current case appeared to indicate an autosomal recessive disease. Unfortunately, we were unable to examine the case further because of the patient’s death.
Conclusion
This case report yielded two main findings in a patient with HHT. First, we detected a missense variant of GDF2 NM_016204.4:c.352A > T(p.Ile118Phe), located within a conserved protein region and predicted as neutral polymorphism according to gnomAD; however, the patient’s BMP-9 levels were markedly decreased, suggesting that this variant may interfere with the expression of the protein. Second, no pathological mutations were found in ENG, ACVRL1, or SMAD4, as the predominant genes involved in HHT, and only one variant was identified in GDF2, a gene previously associated with HHT. Further research is needed to verify the correlation between the GDF2 variant and the pathogenesis of HHT.
Footnotes
Acknowledgement
We thank the patient for providing valuable information.
Author contributions
Qiang Gong conceived and designed the study. Le Ma analyzed and interpreted the data and wrote the manuscript. Xi Peng provided the patient’s study material. All authors read and approved the final manuscript.
Data availability
All data analyzed or generated during the study are included in this published article.
Declaration of conflicting interests
The authors declare that there is no conflict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.