
Abstract
Over the past two decades, non-alcoholic fatty liver disease (NAFLD) has become a leading burden of hepatocellular carcinoma and liver transplantation. Although the exact pathogenesis of NAFLD has not been fully elucidated, recent hypotheses placed more emphasis on the crucial role of the gut microbiome and its derivatives. Reportedly, microbial metabolites such as short-chain fatty acids, amino acid metabolites (indole and its derivatives), bile acids (BAs), trimethylamine N-oxide (TMAO), and endogenous ethanol exhibit sophisticated bioactive properties. These molecules regulate host lipid, glucose, and BAs metabolic homeostasis via modulating nutrient absorption, energy expenditure, inflammation, and the neuroendocrine axis. Consequently, a broad range of research has studied the therapeutic effects of microbiota-derived metabolites. In this review, we explore the interaction of microbial products and NAFLD. We also discuss the regulatory role of existing NAFLD therapies on metabolite levels and investigate the potential of targeting those metabolites to relieve NAFLD.
Introduction
Since the 1980s, metabolic diseases like non-alcoholic fatty liver disease (NAFLD) have been predominant worldwide due to improved economic status and the generally accepted ‘westernized lifestyles’, characterized by high-fat and low-fiber diets, sedentary habits, and circadian oscillations.1–3 NAFLD is defined as overall >5% hepatocytes steatosis demonstrated by radiography or histology in the absence of excessive alcohol intake and is strongly linked to metabolic disorders such as type 2 diabetes mellitus (T2DM), hyperlipidemia, and obesity.4–6 Meta-analysis projected the overall prevalence of NAFLD to be 25% globally. 7 Moreover, recent data indicated a 25.28–33.90% increase in Asia per 5 years between 1999 and 2017. 8 Despite its substantial burden on public health, the exact mechanism of NAFLD remains largely unknown. The gut microbiome and its metabolites are so in the spotlight.
The gut contains a diversity of microorganisms and harbors more genes than human parenchymal cells by approximately 150-fold. 9 Technical and methodological limitations used to be obstacles in microbiota research. However, with the advent of the ’omics era, experiments on microbiota composition at low levels like strains and species are feasible. Basically, four types of bacteria, Firmicutes, Bacteroidetes, Proteobacteria, and Actinobacteria, account for nearly 90% of gut flora at the phyla level. 2 And the gut microbiome is dynamically perturbed by environmental and host factors, as changes were observed in autoimmune and metabolic diseases.10,11 For instance, studies found that the abundance of Proteobacteria and Ruminococcus increased in NAFLD, while the latter was associated with fibrosis (⩾stage 2).12,13 Further research grouped 171 NAFLD patients and revealed that Veillonellaceae were relevant to fibrosis in non-obese patients, while Ruminococcaceae are associated with obese ones. 14 Indeed, aside from bacterial signature, virome and mycobiome profiles also changed significantly. Fecal viral diversity and bacteriophage proportion decreased while Candida albicans concentration increased in NAFLD patients with advanced fibrosis.15,16 However, controversial results are occasionally issued, which is partially due to the sophisticated shaped microbiome by genetics, 17 diets, drugs, 18 circadian rhythms, 19 and physical activities. 20
Given the high heterogeneity and variability of the microbiome, increasing research has shed light on microbial metabolites. Ample evidence21,22 supports the vital role of microbe-derived metabolites, including short-chain fatty acids (SCFAs), amino acid metabolites, bile acids (BAs), trimethylamine N-oxide (TMAO), and endogenous ethanol, in modulating intestinal microbiome and pathogenesis of NAFLD. In this review, we discuss several metabolic productions of gut bacteria and examine their links with NAFLD. Moreover, we explore how existing interventions for NAFLD regulate microbial metabolites and investigate novel approaches to treat NAFLD by modifying those metabolites.
NAFLD and gut microbiome
NAFLD refers to a spectrum of liver disorders with hepatic steatosis, ranging from simple steatosis to non-alcoholic steatohepatitis (NASH) with or without cirrhosis. 6 Although genome-wide association studies have identified mutations in patatin-like phospholipase domain-containing protein 3 and transmembrane 6 superfamily 2 in NAFLD, 23 the exact mechanism of it remains unknown. A widely accepted theory is the ‘multi-hit’ hypothesis that contemplates lipid accumulation, oxidative stress, endoplasmic reticulum (ER) stress, insulin resistance (IR), gut microbiota dysbiosis, and inflammatory response.24–26 Moreover, as an indicator of deteriorated prognosis, fibrosis is associated with a higher mortality rate in patients with NAFLD, particularly those with advanced (stage 3 and stage 4) fibrosis. 27 Therefore, lipid metabolism regulation, inflammation relief, and fibrosis remission are critical determinants for NAFLD management (see Figure 128–30).
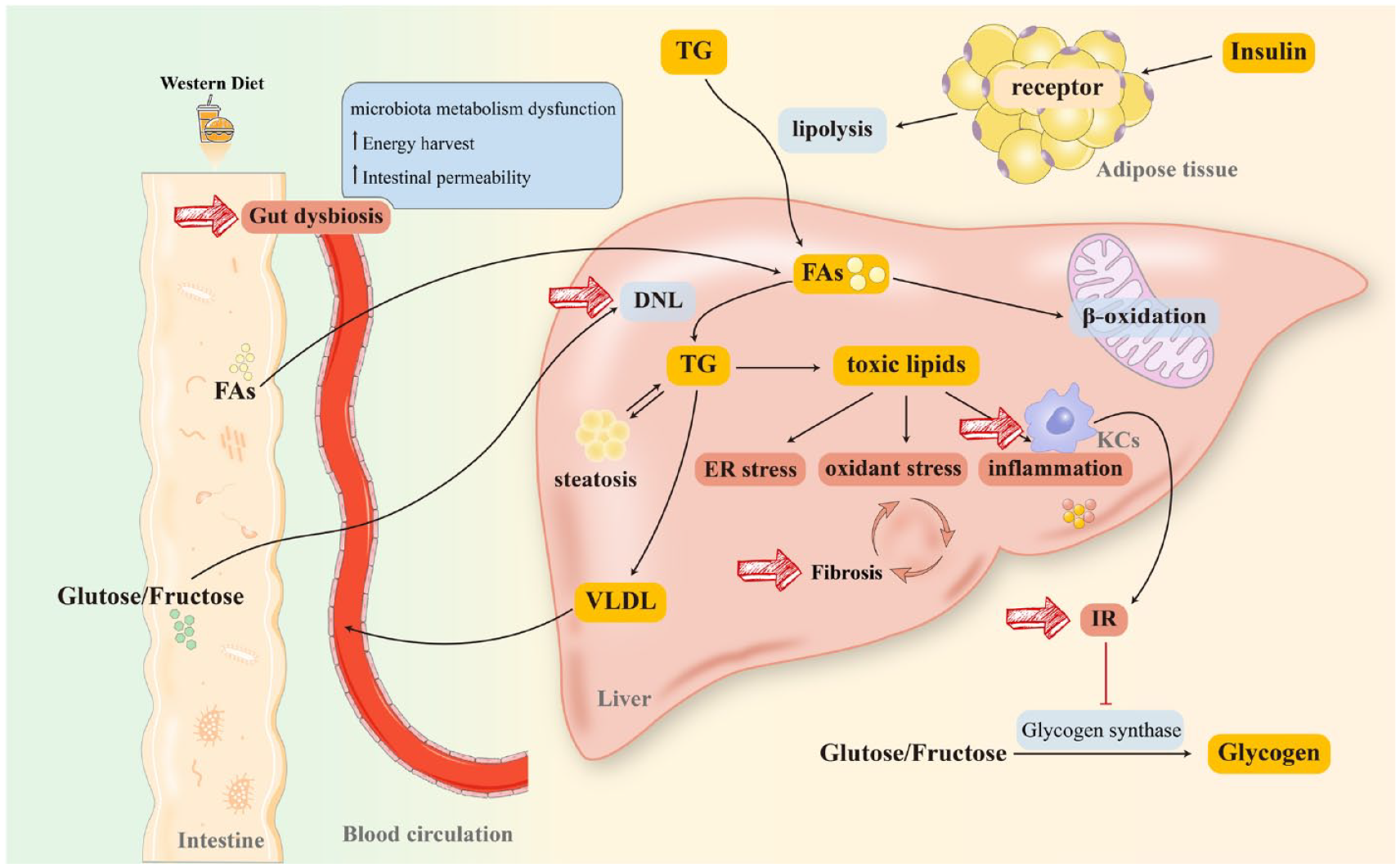
Pathogenesis and therapeutic targets of NAFLD. Long-term consumption of a WD will substantially reprogram the intestinal microbiome. Furthermore, FAs from diet or lipolysis of TG (stimulated by the combination of insulin and receptor on lipocytes) are transported to the liver through blood circulation. As a critical element in initiating NAFLD, IR has been hypothesized to connect with lipid intake. Moreover, IR activates KCs and impairs glycogen synthase, more dietary glucose and fructose reach the liver, resulting in massive FAs accumulation in the liver. A proportion of FAs participates in β-oxidation in mitochondria and peroxisomes to produce energy, while the rest stimulates DNL to synthesize TG. TG is degraded in two ways: (1) being transported to peripheral organs in the form of VLDL and (2) producing lipotoxic lipids (diacylglycerols, ceramides, lysophosphatidyl choline species, cholesterol) that induce mitochondrial dysfunction, ER stress, and inflammatory responses, promoting fibrosis to exacerbate NAFLD/NASH. Diet-induced gut dysbiosis also increases energy harvest and intestinal permeability, as well as perturbs microbiota metabolism, leading to NAFLD progression and pathogenesis. Together, gut dysbiosis, DNL, IR, KCs activation, and fibrosis are all plausible targets for NAFLD management.
As aforementioned, the bidirectional relationships of the gut ecosystem and liver are imperative both physiologically and pathologically. Generally, the liver receives rich nutrients, microbial metabolites, and subproducts from the intestine and secretes bile into the duodenum. 21 An integrated gut barrier also protects against toxins to maintain internal homeostasis. This gut-liver axis is regulated and stabilized by a complex network of metabolic, immune, and neurosecretory interactions between the gut, microbiota, and liver. Disruption of this equilibrium may lead to gut dysbiosis and liver injury.
Gut dysbiosis is characterized by decreased microbiome richness and diversity, as well as a compromised intestinal barrier.31,29 Microbiome dysbiosis has been proposed to exacerbate NAFLD in three ways: enhancing energy harvest, increasing toxic metabolic byproducts, and provoking hepatic inflammation.32,33 High-fat diets (HFDs) disrupt the gut barrier, facilitating the translocation of bacterial derivatives into the blood. 29 Molecules like lipopolysaccharide (LPS) subsequently activate pathogen-associated molecular patterns or damage-associated molecular patterns signaling pathways, contributing to NAFLD progression. 34 Nevertheless, NASH advanced independently of gut barrier impairment in some cases, indicating that microbial outputs other than LPS were involved. 35
In this way, microbial metabolites came to light and attracted much attention. For instance, microbiota-derived molecules have been reported to regulate lipid metabolism, 36 hepatic inflammation,37,38 and liver fibrosis 23 via multiple signaling pathways. According to recent findings, 12 those metabolites have been identified as mediators between microbiota and the steatosis phenome, highlighting their relevance to NAFLD remission.
Microbiota-derived metabolites
Spatially segregated from the intestine by the mucus layer, gut microorganisms contact the host through their metabolic outputs. 39 Fermentation of substrates (like carbohydrates and proteins) yields various metabolic products with intricate functions (Figure 2). 40 The liver degrades metabolites and toxic elements from the intestine. In general, the dynamic wiring of the liver, gut, and metabolites is essential for host metabolism. Below, we detailed these microbial metabolites and their relationships with NAFLD.

Dialogue between gut microbial metabolites and host metabolism. Metabolites derived from gut microbiota contribute to metabolic and endocrine homeostasis in a wide range of organs, including the liver, adipose tissue, pancreas, and brain. SCFAs are critical regulators of lipid metabolism, insulin secretion, satiety, and inflammation by activating GPRs. In addition, SCFAs reduce luminal PH to suppress TPH and redirect tryptophan degradation toward indole. Since indole and its derivatives possess superior properties to inhibit inflammation and lipogenesis compared to other tryptophan compounds like serotonin and kynurenine. As for the BAs, they affect host metabolism and liver fibrosis via an array of signaling pathways, including the hepatic and intestinal FXRs, as well as the TGR5. Aside from that, TMAO and endogenous ethanol contribute to liver inflammation.
Short-chain fatty acids
Among the metabolites, SCFAs were the most extensively studied in NAFLD. 41 SCFAs are typically generated by colonic dietary fiber (DF) fermentation, while those protein-originated account for 17–38%. 42 The reduction of SCFAs has been correlated to lipid dysmetabolism and liver steatosis in high-fat diet (HDF)-induced rodent models, suggesting a plausible target to alleviate NAFLD.43,44 Reportedly, SCFAs exert pleiotropic effects in NAFLD, the majority of which are mediated by G protein-coupled receptors 41 (GPR41)/free fatty acid receptor 3 (FFAR3), GPR43/FFAR2. 45 In general, acetate, propionate, and butyrate account for ⩾95% of SCFAs in a ratio of 3:1:1. 46 Still, the interplay and affinity between each SCFAs and host signaling pathways differ.
Acetate accounts for the vast majority of SCFAs, and is reported to potentiate insulin secretion and β-cell proliferation.47,48 In an HFD-induced mice model, sodium acetate administration suppressed fatty acid synthase (FASN) expression, lowered weight gain, and hepatic triglyceride (TG) accumulation, consistent with the increased acetate-producing Desulfovibrio vulgaris abundance. 49 Olaniyi et al. 50 and Aoki et al. 51 further reported that acetate enhanced insulin sensitivity via suppressing histone deacetylase (HDAC) and activating FFAR2 in mice with diet-induced liver steatosis. Meanwhile, after rectal injection of sodium acetate 52 and a mixture of SCFAs, 53 circulating peptide YY (PYY) and glucagon-like peptide-1 (GLP-1) levels increased in obese individuals. Interestingly, other experiments suggested that acetate contributed to glucose-stimulated insulin secretion (GSIS), weight gain, and hepatic TG accumulation by stimulating parasympathetic activity. 54 Chambers et al. 55 attributed this discrimination to the different properties of acyl-CoA synthetase short-chain family members (ACSS): acetate activated ACSS2 and produced acetyl-CoA for de novo lipogenesis, while ACSS3 converted propionate into propionyl-CoA for mitochondrial respiration and tricarboxylic acid (TCA) cycle.56,57 Isotope tracing indicated that exogenous acetate was used for acetyl-CoA and malonyl-CoA synthesis, promoting lipogenesis and GSIS.57,58
Propionate has been shown to decrease liver and plasma fatty acids (FAs) levels, stimulate GLP-1 and PYY release, improve IR, and regulate energy expenditure through the GPR41 pathway. 59 Furthermore, Ldlr−/− plus HFD-treated mice exhibited a decreased hepatic collagen content following propionate treatment. 60 A prospective study 55 enrolled 18 NAFLD volunteers being treated with either inulin-propionate ester (IPE; 20 g per day) or inulin for 24 weeks and found that intrahepatocellular lipid was substantially less in the IPE group, further supporting the effects of propionate on preventing liver fat accumulation. Reportedly, administration of IPE significantly ameliorated hepatic steatosis and circulating low-density lipoprotein cholesterol (LDL-C), while administration of propionate alone did not show such benefits. The author attributed this discrepancy to inulin (which slows down propionate degradation) and noted that propionate works in the colon rather than the proximal small intestine. 61 Nevertheless, low-dose propionate (0.5–2.0 g/kg) increased glucagon secretion and fatty acid binding protein 4 expression, reducing insulin sensitivity. 62 Despite the conflicting results, more studies tend to believe that colonic fermentation and high propionate concentration (>4% w/w) could improve NAFLD. 63
Colonic epithelial cells primarily utilize butyrate for adenosine 5′-triphosphate production, resulting in a surplus of cAMP promoting intestinal gluconeogenesis and fatty acid oxidation (FAO).64,65 Dai et al. 44 demonstrated that maternal sucralose feeding induced NAFLD in offspring mice with decreased butyrate-producing Clostridium butyricum. In rodent models, butyrate66,67 and butyrate-producing probiotics 68 have been reported to ameliorate steatosis. Sodium butyrate administration in HFD-fed mice decreased liver steatosis by activating the p-AMP-activated protein kinase (AMPK)/p-acetyl-coenzyme A carboxylase (ACC) pathway and restored hepatic GLP-1R levels by suppressing HDAC2. 69 Butyrate administration (by oral gavage for 2 weeks) upregulated thermogenesis-related uncoupling protein 1 (UCP1) and FAO-related carnitine palmitoyltransferase-1 alpha in adipose tissue. 70 In addition, butyrate reduced hepatic collagen content through inhibiting toll-like receptor 4 (TLR4) expression and Kupffer cells activation, 71 as well as the non-classical transforming growth factor β signaling pathways. 72 It also prompted M2 macrophage polarization, 46 revealing the anti-inflammatory property of butyrate. Likewise, butyrate improved intestinal barrier integrity and prevented microbiota-derived toxin translocation. 73
SCFAs displayed versatile effects on glucolipid metabolism, inflammatory response, and liver fibrosis regulation. However, consistent results were not always guaranteed. There are three possible interpretations of these discrepancies. In the first place, some studies failed to ensure that SCFAs are fermented in the colon, resulting in improper links between their effects and diseases. In addition, the affinity of different SCFAs to GPRs differs between humans and mice, resulting in compromised translatability of animal findings. 45 Furthermore, as a critical energy source for ceco-colonic epithelium, nearly 95% SCFAs are absorbed and utilized by the colon, while the remaining 5% are excreted in the stool.74–76 By contrast, in the current research, SCFAs are typically measured in feces or cecum content rather than blood samples from the portal vein, indicating that the change in SCFAs might not have been accurately assessed. Indeed, the ratio of fecal acetate:propionate:butyrate changed in HFD-induced mice as compared to the normal diet group (74:21:5 versus 58:26:16). With this ratio restored, steatosis was significantly relieved, 41 implying that the modification of SCFAs ratio would be more reliable in NAFLD treatment.
Amino acid metabolites
Amino acids are degraded and metabolized into bioactive molecules, exerting diverse properties in metabolism. 77 For instance, as a vital aromatic amino acid (AAA), intestinal tryptophan metabolism consists of three routes. The kynurenine and serotonin [5-hydroxytryptamine (5-HT)] pathways are host dominated, while the indole pathways are microbial mediated.46,78 Reportedly, microbiota-derived indole and 5-HT improved hepatic steatosis and gut-barrier function. 79
Indole and its derivatives, including indole-3-propionic acid (IPA), indole-3-aldehyde (I3A), indoleacrylic acid, and indole-3-acetic acid (IAA), are aryl hydrocarbon receptors (AhRs) agonists to stimulate CD4+ T cells differentiation toward regulatory T cells and regulate lipid metabolism. 79 In inflammatory bowel disease (IBD), indole alleviated intestinal inflammation by upregulating interleukin 22 (IL-22) and downregulating interferon gamma in lamina propria mononuclear cells. 78 Among NAFLD patients, the plasmatic indole concentration was inversely correlated to hepatic fat content and pro-inflammatory macrophage polarization. 37 In addition, intraperitoneal injection of IAA (50 mg/kg) decreased lipogenesis-related stearoyl-CoA desaturase and peroxisome proliferator-activated receptor γ (PPARγ) expression, increased antioxidative superoxide dismutase and glutathione (GSH) levels in HFD-induced C57BL/6 mice. 24 Furthermore, IPA improved intestinal barrier integrity through upregulating zonula occluden-1 and occludin. It also alleviated liver steatosis by suppressing nuclear factor κB (NF-κB) and its downstream pro-inflammatory cytokines, such as tumor necrosis factor α (TNF-α), IL-1β, IL-6. 80 However, Knudsen et al. 81 reported that indole eliminated hepatic M1 macrophages and ameliorated liver function independent of improving lipid metabolism and intestinal barrier function. The discrepancy might be attributed to the relatively low content (0.5 mM) and the method of administration (by drinking water).
While merely 5% of 5-HT is microbiota-derived, it is believed to be extensively critical. 82 It consists of two types, central serotonin suppressed appetite and peripheral 5-HT was correlated with obesity, as tryptophan hydroxylase 1 (a rate-limiting enzyme in peripheral 5-HT synthesis) KO promoted thermogenesis and reduced fat deposition in HFD-treated mice.83,84 Reportedly, intraperitoneal kynurenic acid injection (5 ml/kg) decreased pro-inflammatory cytokines and upregulated UCP1 by activating GPR35 in mice adipose tissue. 85 Pyun et al. 86 demonstrated that kynurenic acid mitigated ER stress and hepatic steatosis in NAFLD. Notably, a cross-sectional study of 233 obese subjects identified elevated serum tryptophan, tyrosine, and phenylalanine as risk factors for NAFLD. 87 This increase may be partially due to impaired intestinal and hepatic catabolism, and the correlation between tryptophan metabolites and NASH was not reported there.
Tyrosine was metabolized into acetyl-CoA to stimulate lipid synthesis and intestinal inflammation. 88 A study enrolled 165 NAFLD patients and observed an elevated 3-(4-hydroxyphenyl) lactate level. It is a metabolite of tyrosine and is significantly associated with liver steatosis and fibrosis. 23 In contrast, plasma glycine decreased in NASH, supplementation of it (at a minimum dose of 27 mg/kg per day) and DT-109 (a tripeptide comprising glycine and leucine) elevated FAO with substantial improvement in hepatic steatosis. 77 Furthermore, microbes in steatosis subjects degraded phenylalanine into phenylacetic acid, reducing insulin response and promoting hepatic steatosis. 12
Branched-chain amino acids (BACCs), including valine, leucine, and isoleucine, can be converted into valerate, isovalerate, and isobutyrate by microbiota. 82 BACCs and their metabolites accumulated in NAFLD and NASH, promoting IR through stimulating gluconeogenesis and mechanistic target of rapamycin complex 1 (mTORC1).89,90 Meanwhile, Prevotella copri and Bacteroides vulgatus (proved to increase in patients with IR) were associated with BACC synthesis. 91 For instance, 3-hydroxybutyrate (3-HIB), a valine metabolite, activated endothelial FAs transport and stimulated lipid deposition in muscle. 92 To summarize, amino acid metabolites exert favorable or adverse effects to either improve or exacerbate NAFLD, and further studies are necessary to unravel their vast functions.
Bile acids
As essential metabolic components, BAs bridge the intestine and the liver. Approximately 95% of them are reabsorbed into the liver in the terminal ileum, a process known as enterohepatic circulation. 93 The remaining 5% of primary BAs, such as cholic acid (CA), chenodeoxycholic acid (CDCA), and corresponding taurine- or glycine-bound derivatives, are further converted to secondary BAs [such as deoxycholic acid (DCA), lithocholic acid (LCA) and ursodeoxycholic acid (UDCA)]. 94 The process includes deconjugation, dehydroxylation, oxidation, or epimerization by gut flora. 95 Moreover, bacteria modified BAs into hydrophobic types (e.g. LCA and DCA) to facilitate their excretion in stool, which stimulated hepatic BAs de novo synthesis and lowered the cholesterol pool by activating the hepatic farnesoid X receptor (FXR).96–98 Reportedly, BAs-modifying bacteria (Ruminococcaceae, Lachnospiraceae, and Blautia) decreased in NAFLD, resulting in metabolic disturbances and chronic inflammation.14,34
BAs perform various functions, primarily through the FXR and takeda G protein-coupled receptor 5 (TGR5) pathways. As the first BAs receptor discovered, FXR enriches the liver, intestine, and kidney, promoting robust fibroblast growth factor 19 (FGF19) or FGF15 (a mouse ortholog of human FGF19) secretion. 99 Indeed, studies have reported the lipid homeostasis regulatory role of FGF15/19 and FXR pathways. In HFD-induced mice models, FGF15 KO accelerated liver steatosis by upregulating PPARγ2 expression. 100 Moreover, the variant of FGF19 downstream klotho beta (KLB), which lead to KLB inhibition, stimulated NAFLD progression through elevating lipogenesis enzymes (p62 and acyl-CoA oxidase 1 (ACOX1)) and pro-inflammatory cytokines (IL-1β and TNF-α) expression. 101 In some research, FXR KO resulted in steatosis deterioration, 102 while other studies correlated FXR knockdown with preventing diet-induced obesity and NAFLD. 21 Researchers attributed these conflicts to various functions of different FXRs, the downstream signaling of hepatic FXR is the small heterodimer partner (SHP) instead of FGF19 in the intestine. Selective ablation of intestinal FXR improved glucose tolerance, 103 while activating intestinal and blocking hepatic FXR pathways accelerated lipid synthesis. 104 TGR5 is predominantly expressed in non-parenchymal cells and activated by BAs selectively (LCA > DCA > CDCA > CA). Besides, BAs stimulated GLP-1 secretion in intestinal L cells, promoted brown adipose tissue and skeleton thermogenesis via TGR5 activation, and increased thyroid hormone by activating type 2 deiodinase, fostering energy expenditure.9,40,59,105
Similarly, the affinity of BAs for FXR varies. Antagonistic DCA increased in NAFLD while agonistic CDCA declined, thus diminishing the protective effect of FXR pathways. 106 In obesity and NASH, elevated DCA level induced TLR2-mediated senescence-associated secretory phenotype and cyclooxygenase 2 expression in hematopoietic stem cells, leading to DNA damage and hepatic carcinogenesis. 107 Hydrophobic LCA also contributed to reactive oxygen species (ROS) production and inflammatory pathways activation. 40 Consistently, hydrophobic, cytotoxic, and 12α-OH BAs (DCA, TDCA, and GDCA) increased in NAFLD and correlated with fibrogenesis.34,108 TCA content in the Western Diet (WD)-treated group was 50-fold higher than in the normal chow group, and was reported to reduce gluconeogenesis.109,110 On the other hand, TCA upregulated pro-inflammatory genes and interfered with the very LDL secretion. 111 CA feeding was also reported to activate hepatic FXR and its downstream SHP and MAF bZIP transcription factor G to inhibit TG accumulation in WD-induced mice. Moreover, Clifford et al. 112 demonstrated that FXR agonists reduced CA and TCA levels and highlighted the discrepancy between the hepatic and intestinal FXR.
Over the past two decades, BAs mimetics that target FXR and TGR5 have been proved to alleviate chronic metabolic and inflammatory disorders in NASH. 105 Recently, a novel concept of ‘microbially conjugated bile acids’ (MCBAs) has garnered considerable attention. As compounds synthesized by conjugating amino acids (phenylalanine, leucine, and tyrosine) and BAs, MCBAs appear to be potent FXR agonists, 95 providing a brand new target for NAFLD treatment.
Trimethylamine N-oxide
TMAO is derived from dietary precursors (phosphatidylcholine, choline, and carnitine) through gut microbial enzymes and hepatic flavin-containing monooxygenase-3. 113 Therefore, microorganisms with TMA-lyase raised TMAO levels.74,114 In the POUNDS Lost trial, Heianza and his colleagues assessed the change of blood TMAO concentration in 504 overweight/obese, and noted that TMAO was inversely associated with insulin sensitivity. 113 TMAO has been widely reported to stimulate foam cells and atherosclerosis plaque formation in artery diseases. 115 In NAFLD, serum TMAO levels were positively related to steatosis grade. 116 Nevertheless, according to a metagenomic study, 12 plasma TMAO, acetate, and phenylacetylglutamine increased in non-steatotic patients with high microbiome richness. Together, further experiments are needed to clarify where the controversy stems.
Ethanol
Unlike exogenous alcohol, endogenous ethanol is generated by the microflora in the colon, leading to liver injury and steatosis. In NASH patients, the circulating ethanol content, the abundance of alcohol-producing bacteria (Escherichia coli), and the expression of hepatic ethanol metabolism-related signaling pathways were elevated.34,117,118 The mice treated by gavage with high alcohol-producing Klebsiella pneumoniae (isolated from the stool of a patient with the bacterial auto-brewery syndrome) displayed upregulated cytochrome P450 2E1 (CYP2E1) and lipogenesis-related genes, resulting in steatosis. 119 Furthermore, endogenous alcohol contributed to mitochondrial dysfunctions. 120 Consequently, therapies targeting microbiome dysbiosis or converting endogenous ethanol to benign molecules like acetate would be plausible.
Other metabolites such as folate and vitamins have also been reported to regulate enterocyte, hepatocyte, and adipocyte metabolism in an epigenetic manner. 2 In addition, neuromodulatory metabolites like gamma-aminobutyric acid (GABA) and catecholamines are critical components of the gut–brain axis, regulating energy metabolism in different dimensions. 82 Silva et al. 121 reported a metabolome signature characterized by decreased butyrate and taurine, as well as increased acetate, tyrosine, and phenylalanine levels in high-fructose diet-fed mice. All of these changes are closely linked to the pathogenesis of NAFLD. Overall, restoring the equilibrium disorder of microbial metabolites may be feasible for NAFLD treatment.
Therapies
To date, no drug has been approved by the European Medicines Agency to be specifically tailored for NAFLD or NASH. 110 In general, the first-line therapy is lifestyle modification, including diet adjustment (adopting a Mediterranean Diet, restricting fructose and alcohol intake) and regular physical exercise. 71 Nevertheless, less than 10% of patients attained their weight loss target, strengthening the significance of developing pharmacological and other novel therapies to induce clinical remission. 122 As stated previously, numerous microbial metabolites improve or exacerbate NAFLD. Here, we detail how these metabolites are affected by NAFLD interventions, including lifestyle modification, drugs, fecal microbiota transplantation (FMT), and bariatric surgery, and try to elucidate how these alterations would impact NAFLD.
Lifestyle
Diet
Diet structure (the proportion of carbohydrates, proteins, and lipids) determines the quantity and the type of substrate being fermented. 42 A diet meeting guideline recommendations is typically high in fiber/carbohydrate, low in fat, sugar, and animal protein. Furthermore, Prevotella were linked to fiber-rich diets, while Bacteroides were related to protein-rich diets. 4 Thus, an adjusted diet might be plausible to correct microbiome dysbiosis. In fiber-deficient diets, bacteria degraded mucins to overgrow and attenuated the mucus layer, elevating intestinal permeability and circulating endotoxins.123,124 By contrast, fiber-rich meals promoted BAs, SCFAs, and IPA production. As reported, Macroalgae Laminaria japonica 125 increased fecal SCFAs with elevated SCFAs-producing Phascolarctobacterium and Allobaculum abundance, downregulated sterol regulatory element-binding protein-1c (SREBP-1c) and upregulated FAO-related ACOX1. In HFD-fed mice, tomato powder 126 reduced Clostidium spp. (with 7α/β-dehydroxylase producing DCA) and attenuated NAFLD. Interestingly, in WD-fed mice fed with oat bran and rye bran (both were 10% of food for 17 weeks), serum parameters of liver injury, inflammation, and gut barrier integrity were improved, independent of decreased liver steatosis. 127 However, this study did not further investigate the effects of oat and rye in a higher concentration. 128
Diet adjustments effectively relieved hepatic steatosis, lipid dysbiosis, and even fibrosis in NAFLD. Unfortunately, the clinical target is often difficult to achieve due to the difficulty of strict implementation. Moreover, diet modification is less effective in individuals with a low-gene content microbiome. 4 Overall, further investigation is necessary to explore the underlying molecular mechanism and develop a personalized diet suitable for each NAFLD patient.
Exercise
Exercise has been reported to attenuate IR and suppress FAs de novo synthesis independent of weight loss. 71 According to the American Gastroenterological Association, 150–300 min of moderate-intensity or 75–150 min of vigorous-intensity aerobic exercise per week is recommended, especially for NAFLD patients with high body mass index (BMI). 129 Evidence showed that exercise increased lipophage by activating the FGF21-mediated AMPK/unc-51 like autophagy activating kinase 1 pathway in muscle. In addition, the interplay between exercise and gut microbiota has also been demonstrated, and Akkermansia has been observed to enrich well-trained athletes.130,131 Furthermore, increased SCFAs production may be another putative effect of exercise. 91 Yang et al. 132 showed that physical activity elevated plasma acetate content, activated GPR43, and improved IR in the diet-induced T2DM mice model. In addition, SCFAs like acetate enhanced exercise capacity, 133 indicating the close connection between exercise and microbial metabolites.
Circadian rhythm
The circadian rhythm controls energy production and BA metabolism. Rhythm disturbance disrupted cholesterol, lipids, glucose, and amino acid metabolism, a crucial etiology of NAFLD and NASH.3,134,135 During sleep, specific microbial metabolites like ergothioneine 136 were eliminated automatically, reducing ROS accumulation and preventing inflammation. In period (Per) 1/2-deficient mice (mice devoid of the core clock transcription factor), bioactive microbiota products regulated hepatic transcript rhythm, with timed feeding restoring normal perturbation rhythm of metabolite and microbiota. 137 Although how the circadian modifies microbial metabolites and host metabolism remains largely unknown, this research regards normal circadian rhythm as a basis of NAFLD therapy.
Pharmacology
Current pharmacotherapies for NAFLD target mainly hepatic fat accumulation, oxidative stress, fibrosis, and gut. 138 Specifically, most of the gut-directed interventions focus on the microbiome. As the therapeutic role of microbial metabolites has been increasingly reported, we summarize the changes of microbial metabolites in several treatments for NAFLD. (Table 1)
NAFLD interventions regulate gut microbiota and its derivatives.

ApoE, apolipoprotein E; BAs, bile acids; CA, cholic acid; CDCA, chenodeoxycholic acid; DCA, deoxycholic acid; Down, downregulate; FAs, fatty acids; FXR, farnesoid X receptor; Gly-MCA, glycine-β-muricholic acid; HDCA, hyodeoxycholic acid; HFD, high-fat diet; I3A, indole-3-aldehyde; 2-HIB, 2-hydroxybutyrate; IAA, indole-3-acetic acid; IBATi, ileal bile acid transporter inhibitor; KO, knockout; LCA, lithocholic acid; L. reuteri, Lactobacillus reuteri; MCD, methionine-choline-deficient; MTF, metformin; MTZ, metronidazole; NAFLD, non-alcoholic fatty liver disease; OAT, oat bran; OCA, obeticholic acid; PDFF, proton density fat fraction; RYE, rye bran; SCFAs, short-chain-fatty acids; T-βMCA, tauro-β-muricholic acid (T-βMCA); TG, triglyceride; TLCA, taurine-lithocholic acid; UDCA, ursodeoxycholic acid; Up, upregulate; WD, western diet; T-α-MCA, tauro-α-muricholic acid.
Pre- and pro-biotics
Gut microbiome-targeted pre- and syn-biotic therapies have been recognized as efficient interventions to alleviate hepatic ultrasound steatosis grade [odds ratio: 2.40, 95% confidence interval (CI) 1.50–3.84, based on a meta-analysis of 21 trials]. 144 As aforementioned, metabolites such as SCFAs, indole, and IPA are prebiotics to ameliorate steatosis and IR in rodent and human models. Besides, DFs (such as polysaccharides, oligosaccharides, and resistant starches) that can be converted to beneficial metabolites are also prebiotics.42,127 According to another meta-analysis of 185 cohort studies, a DF intake between 25 g per day and 29 g per day would considerably lower body weight and plasma cholesterol, as well as risks of T2DM [relative risk (RR): 0.84, 95% CI: 0.78–0.90] and cardiovascular disease (RR: 0.69, 95% CI: 0.60–0.81). 145 Studies revealed the regulatory role of DF on microbiota metabolism in NAFLD (see Table 1).
Furthermore, studies noted that the fermented position and physicochemical characteristics (solubility, viscosity, and fermentability) of DFs are critical determinants. Increased viscosity would affect BAs reabsorption and intestinal transit time (prolonging gastric emptying time and enhancing satiety). 75 Moreover, fibers with smaller particle sizes produced more SCFAs, probably due to the larger surface area exposed to microbial enzymes. Furthermore, the effects of fiber co-administration are more remarkable. A 3-week randomized controlled trial (RCT) found that wheat bran plus resistant starch boosted acetate and butyrate production more significantly than wheat bran alone. 146 Reportedly, the fermentation of a single fiber slowed down after being mixed up, which enabled fibers to reach the distal colon. 75 In general, approaches can be used to enhance the therapeutic effect of DFs, including molecular structure modification and multi-fiber combinations.
Other prebiotics has also been reported to alleviate steatosis. Xu et al. 79 demonstrated that sulforaphane increased IAA levels, downregulating lipogenic genes through AhR activation. It was also reported that ferulic acid-treated mice with increased plasma I3A levels experienced similar steatosis consolation. 139 Necro X compounds 147 derived from indole improved hepatic steatosis and inflammation in methionine-choline-deficient diet-fed mice through suppressing ROS and TNF-α/NF-κB pathways. In addition, Grape polyphenols, 148 Monascus purpureus-fermented common buckwheat, 109 and inulin 149 all contributed to microbial SCFAs production and liver protection in diet-induced NAFLD mice. Besides, recent clinical trials recruited 98 NAFLD patients and found that Mastiha (2.1 g per day for 6 months) improved the antioxidant and inflammatory status of NAFLD at the gene level, as well as reduced the hepatic proton density fat fraction (PDFF) and stimulated the conversion of CA to DCA in obesity (BMI > 35 kg/m2).140,150
Probiotics of beneficial commensal strains such as Lactobacillus and Bifidobacteriu are widely used. 151 Several probiotics promote GLP-1 secretion, glucose tolerance, satiety, and gut barrier integrity. 152 VSL#3, a prebiotic containing four Lactobacillus and three Bifidobacterium species, has shown significant potential to diminish steatosis and fibrosis in WD-induced fxr-/- mice. As it enhanced butyrate-producing Ruminococcus, Faecalibacterium abundance and refined BAs profile with increased UDCA, decreased DCA and hyodeoxycholic acid. 102 In addition, an RCT 141 of 31 pediatric NAFLD patients revealed that VSL#3 supplementation substantially decreased urinary 2-HIB (derived from valine) and steatosis grade. Meanwhile, Lactobacillus lactis significantly restored SCFAs, BAs, and tryptophan metabolite levels in WD-fed mice. 110 Akkermansia muciniphila and Ruminococcus are mucin-degrading bacteria maintaining gut barrier integrity.9,18,96 Akkermansia muciniphila was demonstrated to treat NAFLD, 153 whereas other studies correlated it with intestinal inflammation in IBD. 18 A recent study combined probiotics (Lactobacillus reuteri, a butyrate-producing Firmicutes), antibiotics (metronidazole, antibiotic against acetate-secreting Bacteroidetes), metformin (MTF) and observed significantly inhibited malondialdehyde and p-AKT/mTOR axis, elevated GSH content and restored fecal SCFAs ratio, almost eliminating hepatic steatosis. 41 To conclude, the regulation of an isolated beneficial microbiota strain failed to ensure an affirmative outcome in the context of complex microbiota–microbiota and microbiota–host interaction.
BA metabolism modification
Drugs to modify BA metabolism have been extensively studied in NAFLD treatment. Indeed, BAs are involved in the dialogs between the host, microbiota, and other microbial derivatives. As aforementioned, the pharmacological effects of FXR have not reached an agreement. Both agonists [obeticholic acid (OCA)] and antagonists (UDCA) have been demonstrated to improve NAFLD. For instance, UDCA elevated BAs synthesis by activating CYP7A1, and decreased LDL-C levels by inhibiting endogenous FXR. 154 Glycine-β-muricholic acid antagonizes FXR as well. It downregulated FAs synthesis-related genes (SREBP1c and FASN) with reduced fecal SCFAs content and upregulated BAs de novo synthesis-related genes (CYP7A1, CYP7B1, and CYP27A1) with increased fecal tauro-α-muricholic acid (T-αMCA) and T-β-MCA. 142 On the other hand, FXR agonist OCA sealed the gut vascular barrier and restored the intestinal microbiome, ameliorating liver steatosis.29,143,155, For instance, the Farnesoid X Receptor Ligand Obeticholic Acid in NASH Treatment trial enrolled 283 subjects with biopsy-ensured NASH, offering OCA or placebo 25 mg/d for 72 weeks, and histology improvement was observed in OCA group (RR: 1.9, 95% CI: 1.3–2.8). 156 Aside from this data, the phase III REGENERATE trial further reported that OCA treatment (25 mg per day) for 18 months ameliorated fibrosis in NASH. 157 Other FXR agonists also showed regulatory effects on BAs and lipid metabolism. GSK2324 112 inhibited TG synthesis-associated genes and reduced intestinal lipid absorption by activating the hepatic FXR signaling pathway. Furthermore, given the correlation between BAs excretion and NAFLD, some drugs are designed to diminish BAs. For example, BAs sequestrant colesevelam upregulated BAs synthesis-related CYP8B1 and decreased cholesterol levels. 158 Ileal bile acid transporter inhibitor also substantially improved gut microbiota α-diversity and relieved steatosis in mice. 93
Nonetheless, a meta-analysis of 76 trials reported that BAs modification therapies (including OCA and UDCA) did not improve NAFLD and elevated risks of adverse results (RR: 1.20, 95% CI: 1.07–1.35). 159 However, this may be partially due to the low quality of trials enrolled and limited observation period, indicating that more refined multi-center RCTs should be conducted.
Insulin regulation
IR is an integral element of NAFLD, characterized by compromised extrahepatic (including adipose tissue and muscle) glucose disposal. Moreover, T2DM is often comorbid with NAFLD and is related to a significant increase in fibrosis (by two times). 30 Anti-diabetes drugs (like MTF, GLP-1, and pioglitazone) are extensively tested in NAFLD. 159 For instance, MTF, an insulin sensitizer, enhanced the abundance of butyrate-producing taxa and stimulated FAO, improving the weight gain and prognosis of NAFLD. 160 Meanwhile, MTF reduced the abundance of Bacteroides fragills, which express both bile salt hydrolase and 7α-hydroxysteroid dehydrogenase to produce conjugated UDCAs.34,40 GLP-1 in NAFLD has been observed to inhibit appetite through the vagus, which diminished in vagotomized rats. Other studies did not report the benefits of GLP-1 in steatosis, possibly due to the downregulation of GLP-1R induced by HFD. 69 Based on the above findings, butyrate restored GLP-1R levels that FAs suppressed. Thus, the combination of GLP-1 and sodium butyrate is worth trying. PPARγ agonists like pioglitazone might be unfeasible given their side effects, including weight gain (at an average of 2.4–4.8 kg), fluid retention, bone loss, and fracture.30,155 Den Besten et al. 161 reported the benefits of moderate PPARγ downregulation. Moreover, it demonstrated that SCFAs decreased TG through liver PPARγ and improved insulin sensitivity by adipose PPARγ. Moreover, lanifibranor, a novel pan-PPAR agonist under clinical trial, has shown significant potential in NASH treatment (NCT03008070).134,162
Fecal microbiota transplantation
FMT is an essential tool to relieve refractory or recurrent Clostridioides difficile infection. It transfers the healthy donor-derived gut flora into the intestine of recipients to restore the normal intestinal microbiome. 163 The transmission of disease susceptibility (NAFLD, obesity, T2DM, IBD) through FMT has been shown in germ-free mice models.34,40 Furthermore, germ-free mice with HFD accumulated less liver lipid than chow-diet-fed mice, suggesting that microbiota and its metabolites are crucial mediators delivering energy between the gut and the liver.34,117
In addition, FMT regulated microbial metabolites to reverse IR and intrahepatic endothelial dysfunction caused by HFD. 21 According to RCTs, the fecal concentration of CA, acetate and butyrate, serum level of GABA, as well as the abundance of acetate-producing Bifidobacterium pseudolongum, butyrate-producing Eubacterium and Lactobacillus spp., and GABA-producing Lactobacillus brevis elevated significantly after FMT, revealing a solid connection between FMT and microbe metabolism.164,165 Consistently, FMT from CD-fed to HFD-fed mice increased cecum butyrate concentration, attenuating hepatic steatosis. 166 Craven et al. 167 administered FMT to 21 NAFLD patients and found that FMT reduced small intestinal permeability with increased gut microbiota richness. However, hepatic PDFF reduction is observed after 6 weeks rather than 6 months, indicating the insufficient certainty and persistence of the effects, so in-depth studies would be necessary to decipher the molecular mechanism. A double-blind RCT investigating the relationship between FMT and microbial metabolites in NAFLD is underway (NCT04465032).
The limitations of FMT include the heterogenic effects of different methods (colonoscopy/enema/naso-duodenal), 18 the risk of infection transmission from donors to recipients, 168 and the short duration. 76 Based on these drawbacks, solutions are proposed, such as adopting a multi-donor strategy 169 to ensure long-lasting effects. Besides, administrating FMT from a metabolite-regulating perspective will provide a new landscape to implement FMT in NAFLD.
Bariatric surgery
Currently, bariatric surgery is the only effective treatment for morbid obesity, yielding rapid and sustained weight loss (15–25%).30,170 NAFLD is a common comorbidity of obesity, affecting up to 80% of the obese. 30 Guidelines indicate that an over 5% weight loss is sufficient to decrease steatosis, while ⩾7% leads to NASH resolution. 129 Consistently, a 10% weight loss in obese women reduced several plasma cytokines concentrations.76,117 Furthermore, a meta-analysis of 32 cohort studies indicated that bariatric surgery promoted steatosis and fibrosis resolution in 66% (95% CI: 56–75%) and 40% (95% CI: 29–51%) NAFLD patients. 171 The mechanism was related to profound alterations in intestinal microbiome composition and metabolic characteristics induced by the surgery.
Seyfried et al. 59 showed that Roux-en-Y bypass surgery (RYGB) altered microbiota composition. Metabolites including BAs, AAAs, cholesterol, and BCAAs, underwent significant changes. The LCA-mediated cholic acid-7-sulfate stimulated GLP-1 secretion. 172 Nonetheless, RYGB failed to alter the microbiome in FXR KO mice models, indicating that the FXR signaling pathway mediates these regulatory effects.9,123 A study enrolled 18 obese patients who underwent laparoscopic sleeve gastrectomy, the surgery attenuated appetite by decreasing the connectivity between precuneus and putamen (both belonging to the reward centers). Furthermore, this ‘disconnection’ was dominated by changed microbial metabolites. 173
Accordingly, bariatric surgery influenced BA metabolism and intestinal microbiota flora composition. 42 It was feasible to treat NASH with improved histology and biopsy-proven activity scores. 5 Bariatric surgery is a promising option to reduce the burden of NAFLD. Nevertheless, it is not recommended in the guideline for NAFLD given its invasive nature. In this regard, more clinical research is urgently warranted, and it may be plausible to consider NAFLD progression as an indicator for bariatric surgery in obesity.
Conclusions
In recent years, NAFLD has been the second leading cause of end-stage liver disease worldwide. 174 The close link between microbial metabolites and metabolic syndrome has been demonstrated in animal and human research, although the fundamental molecular mechanisms have not yet been established. Our review summarizes current findings regarding microbiota-derived metabolites and the challenges we face.
Technology latency is the most critical obstacle. Since the discovery of 16S RNA sequencing, human cognition of the microbiome has undergone enormous changes. A flourishing bioinformatic approach, including metagenomics and metabolomics, has enabled further investigation of gut microorganisms and their metabolites.18,123 As aforementioned, the exact molecular connection between the gut, microbial metabolites, and NAFLD has not been elucidated, highlighting the necessity of technology evolution. Meanwhile, the current monotherapy for NAFLD and NASH has not shown satisfactory results. Moreover, given the ‘multi-hit’ etiology and the complex microbial metabolites, factors predisposing to NAFLD are often mixed and integrated. In light of this, combination therapies appear to be more viable. 155 For instance, the combination of prebiotics, probiotics, and even prebiotics and FMT has been proved to alter metabolites and relieve steatosis more significantly. Notably, medication safety needs to be evaluated more thoroughly. Furthermore, recent studies have provided additional avenues for attenuating NAFLD in a microbial metabolite-dependent manner. A Ketohexokinase inhibitor (PF-06835919), 175 designed to alleviate NAFLD by blocking microbial fructose metabolism, is being evaluated in phase II clinical trials. In addition, unabsorbable drugs protect against gut dysbiosis and liver injury by combining or assimilating toxic metabolites in the gastrointestinal tract.
As a whole, there is sophisticated cross-talk between gut microbiota-derived metabolites and NAFLD, albeit exact causal links have not been identified. Moreover, our understanding remains descriptive and observational, necessitating relentless efforts to bridge the huge gaps. Despite this, the benefits of these metabolites and metabolite-directed interventions have been demonstrated, indicating that the regulation of metabolites and their downstream signaling pathways is of significant potential in treating NAFLD.