
Abstract
Background:
Prevotella copri is the most abundant member of the genus Prevotella that inhabits the human large intestines. Evidences correlated the increase in Prevotella abundance to inflammatory disorders, suggesting a pathobiont role.
Objectives:
The aim of this study was to investigate the phylogenetic dynamics of P. copri in patients with irritable bowel syndrome (IBS), inflammatory bowel diseases (IBDs) and in healthy volunteers (CTRL).
Design:
A phylogenetic approach was used to characterize 64 P. copri 16S rRNA sequences, selected from a metagenomic database of fecal and mucosal samples from 52 patients affected by IBD, 44 by IBS and 59 healthy.
Methods:
Phylogenetic reconstructions were carried out using the maximum likelihood (ML) and Bayesian methods.
Results:
Maximum likelihood phylogenetic tree applied onto reference and data sets, assigned all the reads to P. copri clade, in agreement with the taxonomic classification previously obtained. The longer mean genetic distances were observed for both the couples IBD and CTRL and IBD and IBS, respect to the distance between IBS and CTRL, for fecal samples. The intra-group mean genetic distance increased going from IBS to CTRLs to IBD, indicating elevated genetic variability within IBD of P. copri sequences. None clustering based on the tissue inflammation or on the disease status was evidenced, leading to infer that the variability seemed to not be influenced by concomitant diseases, disease phenotypes or tissue inflammation. Moreover, patients with IBS appeared colonized by different strains of P. copri. In IBS, a correlation between isolates and disease grading was observed.
Conclusion:
The characterization of P. copri phylogeny is relevant to better understand the interactions between microbiota and pathophysiology of IBD and IBS, especially for future development of therapies based on microbes (e.g. probiotics and synbiotics), to restore the microbiota in these bowel diseases.
Introduction
A huge number of studies have reported various types of association between the microbiota composition in health, metabolic disorder and gastrointestinal diseases.1–4 The loss of intestinal homeostasis, named dysbiosis, has been described in different intestinal disorders, including inflammatory bowel disease (IBD) and irritable bowel syndrome (IBS).5,6 IBD, such as Crohn’s disease (CD) and ulcerative colitis (UC), are chronic, relapsing-remitting, gastrointestinal inflammatory diseases, which are associated with various degrees of intestinal damage and intestinal inflammation, due to an excessive and impaired inflammatory response. 7 IBS is one of the common functional gastrointestinal disorders worldwide, characterized by abdominal pain or discomfort bloating associated with altered bowel habits.8,9
Different studies reported a fluctuation in the equilibrium between beneficial commensals and potential pathobionts in gut microbiota as well as alterations in microbial molecular products in IBD and IBS patients.10–14
The role of specific gut bacteria in pathogenesis of these diseases is not exactly known.
Prevotella copri has been reported as the most abundant member of the genus Prevotella that inhabits the human large intestines.15–20 Prevotella has been associated with high fiber-rich diet, such as non-Westernized diet.21,22 Moreover, it has been reported that the increase in Prevotella abundance correlated with glucose metabolism improvement, suggesting a potential beneficial role of these bacteria in human health. 17
However, the increase in Prevotella abundance has been also linked to inflammatory disorders, including periodontitis, bacterial vaginosis, rheumatoid arthritis, ankylosing spondylitis, metabolic disorders and low-grade systemic inflammation, suggesting that at least some strains exhibit pathobiontic properties.23,24 It has been demonstrated that Prevotella exerts its proinflammatory effect by the activation of Toll-like receptor 4 (TLR-4) through lipopolysaccharide (LPS) production25,26 and by the decrease in colonic interleukin-18 expression (IL-18). 27 Moreover, the increment in Prevotella increased intestinal permeability by the production of mucin-degrading enzymes. 28
Although studies of experimental colitis in mice revealed a role of Prevotella in IBD, currently no human studies have confirmed an association between the increase in Prevotella abundance and chronic intestinal diseases.27,29,30
This apparent conflict in Prevotella’s role on human physiology could be resolved by the increase in scientific studies aimed to understand the functionality of Prevotella species/strains. 31 This knowledge could give important information for the future development of therapies based on microbes for the restoring of dysbiotic gut microbiota, especially associated with bowel diseases.
In this study, the genetic diversity and phylogenetic dynamics of 64 P. copri 16S rRNA sequences, selected from a metagenomic database32,33 of stools and biopsies from IBS and IBD patients and from healthy CTRL, have been investigated to comprehend the role of this microorganism in gut pathophysiology.
Materials and methods
Cohort characteristics and sample collection
This study represents a part of the research project (WFR GR-2011-02350817) funded by the Italian Ministry of Health. Specifically, during 2015–2017, we recruited 52 IBD patients at the Department of medicine and gastroenterology of Tor Vergata Hospital (Rome, Italy), 44 IBS patients and 59 healthy volunteers (CTRL) at the Gastroenterology Unit of the Campus Biomedico Hospital (Rome, Italy). This study conforms to the guidelines for STROBE statement. 34
Anthropometric and clinical characteristics of IBD, IBS and CTRL cohorts are reported in Supplemental Tables 1 and 2. More details on the inclusion/exclusion criteria are reported in Lo Presti et al. 33 (2019).
The therapies administered to IBD patients were as follows: 5-aminosalicylic acid or sulfasalazine (46.9%); tumor necrosis factors (TNFs; 12.5%); thiopurine (5.3%); steroids (28.9%); and steroids plus anti-TNF (6.4%). The IBS concomitant therapies were as follows: antispasmodics (16%), antidepressive (7%) and laxatives (18%).
All patients underwent mucosal biopsies during colonoscopy to perform routine histological examinations. In IBD patients, the biopsies in relation to the disease localization (from injured and from macroscopic healthy area, when applicable) were taken in the colon, while in IBS patients and in CTRL the biopsies were taken in the ascending or sigmoid colon. All patients collected a stool sample the day before the colonoscopy preparation or at least 2 weeks after the endoscopic examination. Biopsies were immediately frozen at −80°C, while the stool samples were stored at −4°C up to the time of transport to the hospital and stored at −80°C.
Sequence characteristics
The 16S rRNA-based metagenomics analysis of microbiota of fecal and mucosal samples32,33 revealed the presence of P. copri sequences in 9 IBS, 11 IBD patients and in 15 CTRLs for an overall of 64 sequences of which 31 sequences from stool samples (8 from CTRL, 9 from IBD and 14 from IBS) and 33 from intestinal biopsies (8 from CTRL, 6 from IBD injured area, 5 from IBD healthy area and 14 from IBS).
The 16S rRNA Prevotella reference sequences (157 sequences) were downloaded from the NCBI database (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA391149).
Phylogenetic analysis
The sequences were aligned and manually edited using Bioedit software. 35 Modeltest v. 3.7 36 was used to select the simplest evolutionary model that best fitted the sequence data. To obtain an overall impression of the phylogenetic signal in all the 16S rRNA Prevotella sequences, the likelihood-mapping analysis of 10,000 random quartets has been generated using TreePuzzle 37 as previously described. 38 Phylogenetic reconstructions were carried out using the ML analysis with Phyml v. 3.0 39 with GTR + I + G model of evolution, previously selected. Robustness of the phylogenetic trees was estimated by bootstrap analysis in 1000 replicates (statistically supported bootstrap values, >90%). The software MEGA v. 7 40 was used to calculate the genetic distances among different groups. The genetic distances were calculated using the K2P model with the standard deviation calculated from 1000 bootstrapped replicates among lineages. The comparisons described were all statistically significant (p < 0.05).
Ethics statement
This study was performed within the Research Project ‘Cross Sectional study to evaluate the interactions between gut microflora and immune system at the cross-road of the pathogenesis of Inflammatory Bowel Diseases and Irritable Bowel Syndrome’ (WFR GR-2011-02350817, financed by the Italian Ministry of Health). In this project, each patient who took part gave written informed consent and the study was approved by the local ethics committee (Study Protocol ‘Tor Vergata’ General Hospital GR-2011-02350817 Register of Experiments 44/15; Campus Prot. 24/15 PAR ComEt CBM) as previously reported. 33
Results
Phylogenetic analysis
To assess the phylogenetic map of P. copri in association with sample origin, fecal and mucosal isolates were first grouped together, and then divided into fecal and mucosal groups. The phylogenetic noise of all groups was investigated by means of likelihood mapping and the percentage of dots in the star-like region ranged from 5% to 18.2%. Since none of the groups showed more that 30% of noise, all of them contained enough phylogenetic signal. Maximum likelihood phylogenetic tree applied onto reference and sequences, assigned the 64 reads to P. copri clade, in agreement with the taxonomic classification previously obtained by 16S rRNA metagenomic-based approach (Supplemental Figure 1). 38
Overall group
The computation of the mean genetic distances between P. copri of fecal and mucosal sequences showed slightly higher statistically significant divergence (14.85%) in CTRL, respect to IBD injured (11.73%) and to IBS (10.59%). The mean genetic distance between mucosal CTRL group versus mucosal IBS was 16.76%. The mean genetic distance between mucosal CTRLs versus IBD injured was 15.20%. The mean genetic distance between mucosal IBD injured versus mucosal IBS was 9.94%.
Regarding fecal sequences, the higher divergences were between CTRLs and IBD (11.59%) and between IBS and IBD (11.61%); meanwhile, a slightly lower distance was observed between CTRLs and IBS (10.03%).
Maximum likelihood analysis has been conducted to investigate the intermixing between fecal and mucosal sequences or any classification of P. copri variants (Supplemental Figure 2). A statistically supported cluster (A) and a main clade (B) were found. All the sequences, except four cases located in cluster A, clustered in the main clade. Overall, eight supported internal clusters, composed of intermixed sequences collected from both fecal and mucosal samples, have been highlighted. Globally, 78.6% (22/28) of the mucosal sequences were located inside the eight supported internal clusters, with respect to 87% (27/31) of the fecal sequences.
Fecal group
The maximum likelihood (ML) phylogenetic tree of the fecal group highlighted a main clade within which it is possible to highlight two statistically supported clusters (A and B) and a sub-clade (C) (Figure 1).

The ML phylogenetic tree of Prevotella copri fecal subset. Branch lengths were estimated with the best fitting nucleotide substitution model according to a hierarchical likelihood ratio test and were drawn to scale with the bar at the bottom indicating 0.2 nucleotide substitutions per site. The tree was rooted using the midpoint rooting method. One asterisk along the branches represent significant statistical support for the clade subtending that branch (bootstrap > 90%). Main clade and cluster were indicated.
Cluster A included one sequence from an IBD patient and two from IBS patients. Regarding the clinical characteristic, the IBD patient was affected by Crohn’s disease (CD) with mild endoscopic activity and in clinical remission. One of the IBS patients, belonging to IBS-D subtype, was affected by gastro-esophageal reflux, meanwhile the second, belonging to IBS-C subtype, was affected by Helicobacter pylori-associated chronic atrophic gastritis. Cluster B was composed of two P. copri sequences from the same IBS patient with different branch lengths. Externally, it is possible to highlight the third sequence from the same IBS patient (IBS-C subtype, affected by H. pylori-associated chronic atrophic gastritis).
Finally, the sub-clade C included seven IBD, nine IBS and seven CTRLs sequences. Three statistically supported clusters (I, II and III) were located inside the sub-clade C. Cluster I was composed of four sequences from CTRLs and one from an IBD patient affected by ulcerative colitis (UC) in clinical remission.
Cluster II was composed of five sequences collected from IBS patients: two of them belonged to patients characterized by diarrhea (IBS-D) and two by constipation (IBS-C). Regarding the concomitant diseases, a patient suffered from diverticulitis, and two of gastro-esophageal reflux and of H. pylori-associated chronic atrophic gastritis.
Cluster III included two sequences from IBS, three from a CTRL and two IBD. Among IBS patients, one presented the constipation (IBS-C) subtype and was affected by H. pylori-associated chronic atrophic gastritis, and the second one suffered from diverticulitis and belonged to diarrheal (IBS-D) subtype. The two IBD isolates were from patients both affected by UC without other concomitant diseases.
The left-over sequences were located sparsely or in different not supported clusters with one IBD sequence more externally located, showing a greater divergence of these sequences.
Measuring the mean genetic distances of sequences grouped by the disease/health status (IBD versus IBS versus CTRLs), we observed a mean genetic distance of 11.6% between IBD and CTRLs, of 10.0% between IBS and CTRLs and of 11.6% between IBS and IBD.
The intra-group mean genetic distance increased going from IBS (9.26%), to CTRLs (9.36%) to IBD (13.76%), indicating elevated genetic variability within IBD P. copri sequences and a similar intra-group distance in IBS and CTRLs.
The fecal IBS subset was investigated in detail to define the phylogenetic relationships among P. copri sequences from the different IBS subtypes (Figure 2, panel a).
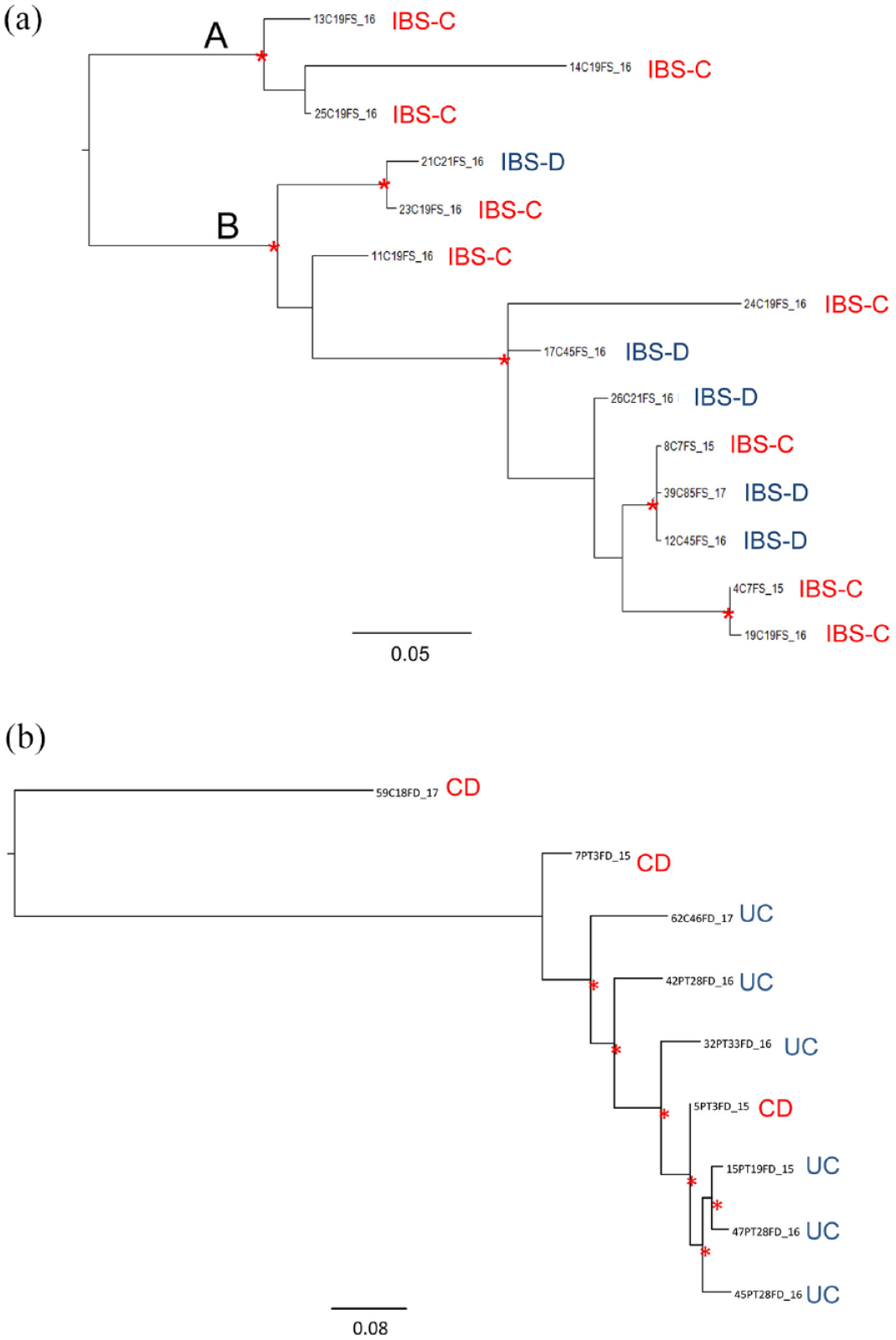
The ML phylogenetic trees of Prevotella copri in fecal subsets. Panel a: IBS subset. Branch lengths were estimated with the best fitting nucleotide substitution model according to a hierarchical likelihood ratio test and were drawn to scale with the bar at the bottom indicating 0.05 nucleotide substitutions per site. The tree was rooted using the midpoint rooting method. One asterisk along the branches represent significant statistical support for the clade subtending that branch (bootstrap > 90%). Main clades were indicated. IBS subtypes were indicated near the tips (red = IBS-C; blue = IBS-D). Panel b: IBD subset. Branch lengths were estimated with the best fitting nucleotide substitution model according to a hierarchical likelihood ratio test and were drawn to scale with the bar at the bottom indicating 0.08 nucleotide substitutions per site. The tree was rooted using the midpoint rooting method. One asterisk along the branches represents significant statistical support for the clade subtending that branch (bootstrap > 90%). The CD and UC isolates were indicated near the tips.
Interestingly, two clades (A and B) were identified. The clade (A) was composed by three IBS-C subtype sequences derived from the same patient and showed different branch lengths. In clade B, the P. copri sequences from IBS-C subtype patients were mainly intermixed with those from IBS-D subtype. Inside this clade, the mean genetic distance of IBS-C and IBS-D sequences was 5.9%. When computing the mean genetic distance including all sequences of this sub-set, a mean value of 6.7% was obtained, between IBS-C and IBS–D. The mean distance intra-group was 7.4% and 4.6% for IBS-C and IBS-D, respectively. A mean genetic distance of 7.8% of divergence was found between clade A and clade B. The computation of the mean genetic distance between clade A and all the IBS-D isolates gave an estimation of 8.1% of divergence.
The ML tree (Figure 2, panel b) of IBD subset showed a main supported cluster with one CD sequence, intermixed with UC sequences; meanwhile, two CD sequences were externally located to the main cluster. Overall, a mean genetic distance of 16.4% was obtained between CD and UC groups. By only investigating the main supported cluster, a mean genetic distance of 3.44% was obtained between CD and UC.
Mucosal group
The ML phylogenetic tree of the mucosal group (Supplemental Figure 3) showed two main clades (A and B), in which a clear separation between IBD (clade A) and IBS (mainly concentrated in clade B) P. copri sequences was evident. All the IBD sequences included in clade A belonged to UC. Interestingly, also the clade B contained the IBD sequences, one of them (6PT13BD_15, derived from an UC patient) representing the outgroup of this clade and other UC isolates were internally located. The sequence from the CD patient (9PT3BD_15) resulted related to two UC, one CTRL and one IBS sequences.
In particular, two sequences collected from ‘macroscopic healthy area’ were strictly related to one from injured area of the same patient (Supplemental Figure 3). This patient showed another mucosal IBD sequence from injured area, which appeared located on clade B in another cluster, suggesting a mild genetic divergence. The mean genetic distance between IBD healthy sequences versus IBD injured was 4.6%.
Phylogenetic analysis of P. copri mucosal sequences from IBS subset showed two supported clades (A and B) (Figure 3, panel a).

The ML phylogenetic analysis of P. copri mucosal sequences. Panel a: IBS subset. Branch lengths were estimated with the best fitting nucleotide substitution model according to a hierarchical likelihood ratio test and were drawn to scale with the bar at the bottom indicating 0.03 nucleotide substitutions per site. The tree was rooted using the midpoint rooting method. One asterisk along the branches represents significant statistical support for the clade subtending that branch (bootstrap > 90%). Main clades were indicated. IBS subtypes were indicated in colors (red, IBS-C; blue, IBS-M; black, IBS-D). Panel b: IBD subset. Branch lengths were estimated with the best fitting nucleotide substitution model according to a hierarchical likelihood ratio test and were drawn to scale with the bar at the bottom indicating 0.02 nucleotide substitutions per site. The tree was rooted using the midpoint rooting method. One asterisk along the branches represents significant statistical support for the clade subtending that branch (bootstrap > 90%). Sequences from the same patients are highlighted by the same color: (i) injured area and (h) macroscopic healthy area.
This analysis included 14 sequences, some of them belonging to the same patient. The ML phylogenetic tree revealed that sequences from different IBS subtypes were intermixed. In clade A, sequences from IBS-D were related to IBS-C and alternating bowel habit phenotype (IBS-M); meanwhile, in clade B, IBS-D sequences were intermixed with IBS-M subtype (Figure 3, panel a). In clade A, sequences from patients with different concomitant diseases were intermixed (i.e. gastro-esophageal reflux and H. pylori-associated chronic atrophic gastritis) with those reporting absences of concomitant diseases. In clade B, the same situation was observed, a sequence from a patient with calcific enthesitis and hypothyroidism was intermixed with two sequences from patient reporting gastro-esophageal reflux and with a patient with absence concomitant diseases. The computation of the mean genetic distance showed that IBS-C group was more distant from IBS-D (8.4%) than from IBS-M (6.2%). The higher value of the mean genetic distance was observed between IBS-D and IBS-M (9.5%). The intra-group mean genetic distance showed the higher value for IBS-D (12.3%), followed by IBS-M (8.5%) and by IBS-C (3.8%).
The ML analysis of IBD sub-set (Figure 3, panel b) showed two statistically supported cluster. The first was composed by one sequence from an UC patient (affected by moderate endoscopic and clinical activity) collected from ‘macroscopic healthy area’. Externally was located a cluster including six UC sequences. These sequences were from ‘macroscopic healthy area’ and from injured area, from three patients characterized by severe endoscopic and clinical activity. The second cluster included three isolates from UC and one from CD. The UC sequences (two sequences were from injured area and one from ‘macroscopic healthy area’) belonged to the same patient characterized by severe endoscopic and mild clinical activity. The CD patient was characterized by mild endoscopic and clinical remission. Both these patients had no other concomitant diseases.
The elaboration of the mean genetic distances between UC and CD including all the sequences was 3.05%; meanwhile, excluding the sequences from ‘the macroscopic healthy area’, a mean value of 8.64% was obtained (Figure 3, panel b).
Discussion
There is a growing number of papers on the importance of the link between Prevotella diversity and human health that are now emerging in the literature. Indeed, this topic is now considered a leading topic in the microbiota literature.20,21,27,41,42
By our phylogenetic approach, applied to 16S-based metagenomics sequences of Prevotella, we obtained the taxon identification up to P. copri species level. In fact, by the ML phylogenetic analysis, the sequences assigned to Prevotella genus were re-assigned to P. copri clade, overcoming the limited identification at genus level of this metagenomic approach.
As previously described, the gut microbiota of IBS patients resulted highly enriched in P. copri respect from IBD and CTRLs. 33 In particular, in our sample set, the ratio of P. copri sequences/nr. patients was 3 for IBS, 1.8 for IBD and 1 for CTRL, suggesting a putative role of P. copri in IBS.
Moreover, by our results, we highlighted the higher genetic variability within IBD P. copri sequences with respect to the lower genetic variability in IBS and CTRLs.
Different studies reported that Prevotella plays a pro-inflammatory role through the activation of TLR-4 by LPS production, resulting in an abdominal pain.25,26 Moreover, it has been demonstrated that high Prevotella levels increase intestinal permeability by the production of mucin-degrading enzymes. 28 Assuming a correlation between isolates and disease grading we investigated the correlation between the sequence clustering and the patients’ clinical information. However, the presence of concomitant diseases in IBD and IBS patients seemed to not influence the distribution of P. copri isolates.
Moreover, we investigated the correlation between IBS subtypes and P. copri sequence variability. By our results, the IBS-C reported the higher intra-group sequence variability, respect the others. Furthermore a higher genetic distance between IBS-C and IBS-D subtypes in fecal samples and between IBS-M and IBS-D subtypes in mucosal ones was reported. Despite in literature has been correlated the increment of Prevotella with the risk of IBS diarrheal phenotype (IBS-D),43,44 in our study, no correlation between isolate variability and IBS subtypes was found. Also for IBDs, the UC and CD phenotypes, the inflamed condition of the tissue and the disease status seemed to not influence the distribution of P. copri.
Our study presents some limitations that could be addressed in future research. First, the sample size should be enlarged to increase the number of P. copri sequences and then the sequence variance to test. Second, the recruitment should include patients with different geographic origin and food habits to investigate the global distribution, the population structure and the relation with diet of P. copri. Third, the investigation should be enlarged to show the correlation between Prevotella species/strains and different disease stages and treatments of IBD and IBS.
Conclusions
In conclusion, unlike patients with IBD, those with IBS appeared to be colonized by different strains of P. copri. The variability of P. copri sequences seemed to not be influenced by concomitant diseases, disease phenotypes or intestinal tissue inflammation. However, in IBS patients, a correlation between isolates and disease grading was observed.
Then, associate the role of single strain in host/microbiota interaction could be useful for the future development of therapies based on microbes (e.g. probiotics and synbiotics), to restore the microbiota in different disorders such as IBD and IBS.
Supplemental Material
sj-docx-1-tag-10.1177_17562848221136328 – Supplemental material for Phylogenetic analysis of Prevotella copri from fecal and mucosal microbiota of IBS and IBD patients
Supplemental material, sj-docx-1-tag-10.1177_17562848221136328 for Phylogenetic analysis of Prevotella copri from fecal and mucosal microbiota of IBS and IBD patients by Alessandra Lo Presti, Federica Del Chierico, Annamaria Altomare, Francesca Zorzi, Giovanni Monteleone, Lorenza Putignani, Silvia Angeletti, Michele Cicala, Michele Pier Luca Guarino and Massimo Ciccozzi in Therapeutic Advances in Gastroenterology
Footnotes
Acknowledgements
The authors would like to thank the Italian Ministry of Health for the funding. The authors would also like to thank Alessandra Avola and Eleonora Cella as contributors to the WFR GR-2011-02350817 Project.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.