
Abstract
Regorafenib is a diphenylurea oral multikinase inhibitor, structurally comparable to sorafenib, which targets a variety of kinases implicated in angiogenic and tumor growth-promoting pathways. Regorafenib was the first agent to positively show significant survival advantage as a second-line therapy in patients with unresectable hepatocellular carcinoma (HCC) who had previously failed first-line treatment with sorafenib. Recent evidence has shown that its antitumor efficacy is due to a comprehensive spectrum of tumor neo-angiogenesis and proliferation inhibition and immunomodulatory effects on the tumor microenvironment, which plays a crucial role in tumor development. This review addresses the rationale and supporting evidence for regorafenib’s efficacy in HCC that led to regorafenib’s approval as a second-line therapy. In addition, we review proof from clinical practice studies that validate the RESORCE trial results. We discuss regorafenib’s potential role in the newly emerging therapeutic strategy based on combination with immune checkpoint blockade and its possible extensibility to patient categories not enrolled in the registrative study.
Keywords
Introduction
Hepatocellular carcinoma (HCC) is the fifth most prevalent cancer and the third leading cause of cancer death globally, with 905,677 new cases and more than 800,000 deaths in 2020, accounting for 8.3% of all cancers. 1
HCC represents nearly 90% of primary liver cancers and is a major global health problem. The incidence of HCC is increasing in most countries and this cancer is currently the preeminent cause of mortality in cirrhotic patients.2–4
Globally, chronic viral hepatitis and alcohol-induced liver disease are the dominant risk factors for HCC development; however, in high-income areas, non-alcoholic fatty liver disease (NAFLD) linked to HCC is rising as a result of the growing prevalence of metabolic disorders.5–7
In contrast, vaccination and treatment for hepatitis B virus (HBV) infection, prevention campaigns for sexual and iatrogenic transmission of HBV and hepatitis C virus (HCV), and the development of effective HCV antiviral drugs are lowering the load of chronic viral liver disease.8–11
Advanced HCC is a lethal malignancy, and, as of 2007, for HCC patients having retained liver function with advanced Barcelona Clinic Liver Cancer (BCLC) stage and who are not eligible for locoregional treatment, the multikinase inhibitor (MKI) sorafenib has been established as the standard of care worldwide. 12
After ten years of dismal results, other agents are currently available as a first-line alternative to sorafenib or second-line after sorafenib failure.13–17
In addition, immune checkpoint inhibitors (ICIs) targeting the programmed cell death receptor-1 (PD-1) and anti-programmed death-ligand 1 (PD-L1) have recently received accelerated approval.14,18,19
In 2016, the randomized, placebo-controlled, phase III RESORCE trial was the first to prove that treatment with regorafenib in patients who previously experienced a failure of first-line sorafenib therapy allowed a significant increase in overall survival (OS), compared with placebo-receiving patients [10.6 versus 7.8 months, hazard radio (HR) 0.63 (95% confidence interval (CI) 0.50–0.79), p < 0.0001], after a decade of failed clinical trials evaluating a wide range of second-line treatments. 15
After registration and its introduction into clinical practice, real-life data have been reported. 20 Recent developments in the systemic treatment strategy for HCC has led to new potential opportunities for the use of regorafenib in combination with other agents.21,22
This review analyzes the primary preclinical data of studies that investigated regorafenib to treat HCC patients and available efficacy and safety data from clinical practice studies. Lastly, we discuss the rationale for its possible use in combination treatment with other agents.
Mechanism of action
Regorafenib (chemical name: 4-4-3-4-chloro-3-trifluoromethylphenylureido-3-fluorophenoxy-N-methylpicolinamide) is a small molecule inhibitor that is one of the biaryl ureic compounds. Regorafenib is an orally available, multitargeted tyrosine kinase inhibitor (TKI) that was developed following a discovery program aimed at enhancing the efficacy of sorafenib; from which it differs only by the addition of a fluorine atom to the central phenyl ring (Figure 1, Panel A). 23
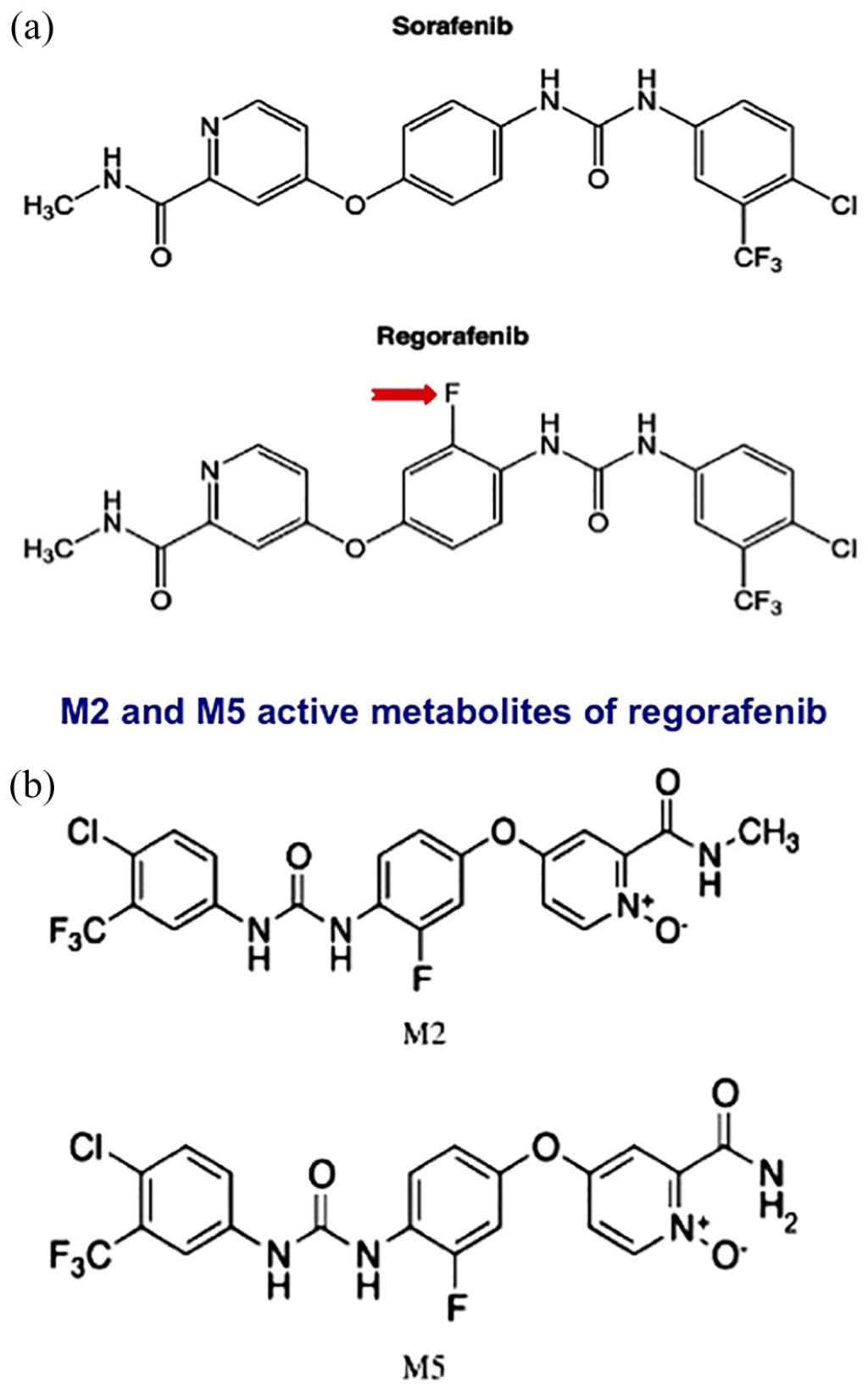
a) Similarly to sorafenib, regorafenib is a bi-aryl urea class of drug. The sole difference between sorafenib and regorafenib is the presence of a fluorine atom in the latter (red arrow). Owing to a mechanism that has not yet been fully defined, this one unique difference produces a wider kinase inhibitory profile. In complement to the targets that are inhibited by sorafenib, regorafenib also blocks the signaling pathway of tyrosine-protein kinase receptor Tie2, the receptor for angiopoietin-2, a pro-angiogenic cytokine. b) M2 and M5 are the main active regorafenib metabolites.
Preclinical studies have demonstrated that regorafenib targets kinases involved in signaling pathways that drive tumorigenesis, cancer spread, and the maintenance of the tumor microenvironment.15,23
Regorafenib features a triple mechanism of action against targets involved in the regulation of angiogenesis, cell proliferation, and tumor stroma. These targets include vascular endothelial growth factor receptors 1-3 (VEGFR1-3), the angiopoietin 1 receptor (TIE2), fibroblast growth receptors (FGFRs), platelet-derived growth factor receptor beta (PDGFR)-β, the oncogenic kinases KIT and RET, and intracellular signaling molecules including those that accelerate fibrosarcoma (c-RAF/RAF-1) kinases, B-RAF, and the BRAF V600E mutant (Figure 2).23–29

Regorafenib can inhibit several molecular pathways by targeting angiogenic, stromal, oncogenic and intracellular kinases. Regorafenib induces M1 macrophage polarization and increases CD8+ T cells proliferation and activation thus also acting on the tumor microenvironment and immunosuppression.
In theory, the dual blockade of the VEGF and TIE2 receptors may significantly enhance the tumor vessel shrinkage effect. Among systemic therapies with anti-angiogenic properties approved for HCC, regorafenib inhibits a broader range of targets (Table 1).
Target structures of systemic therapies with antiangiogenic effects.

Among the approved systemic therapies for HCC with anti-angiogenic effects, regorafenib displays the broadest spectrum of inhibited target receptors.
AXL; tyrosine protein kinase receptor UFO; FGFR, follicular growth factor receptor; KIT, tyrosine-protein kinase KIT; MET, tyrosine protein kinase MET; PDGFR, platelet-derived growth factor receptor; RAF, rapidly accelerated fibrosarcoma kinase; RET, rearranged during transfection protogene; TIE-2, tyrosine protein kinase receptor Tie-2; TKI, tyrosine kinase inhibitor; VEGFR (1-3), vascular endothelial growth factor receptors 1-3.
Some results also suggest that regorafenib demonstrates anti-immunosuppressive properties, as well as promoting anti-tumor immunity. 30 Regorafenib has shown the critical effect of enhancing anti-tumor immunity by modulating macrophages and increasing proliferation and activation of CD8+ T cells (Figure 2). Tumor-associated macrophages (TAMs), a key element of leukocyte infiltration, promote tumor cell growth, development, and migration. 31
The role of TAMs in carcinogenesis is well-documented in several tumor types, including HCC.32,33
Regorafenib inhibits the colony-stimulating factor 1 receptor (CSF1R), which is critical for macrophage differentiation and survival and causes a reduced tumor infiltration of macrophages (Figure 3).27,34,35

Several TME components promote an immune suppressive environment. Regorafenib may modulate an immune-suppressive TME and promote anti-tumor immunity, blocking VEGFRs, TIE2, and CSF-1R.
In agreement, regorafenib has been shown to decrease the infiltration of TAMs, which are crucial for angiogenesis and metastatic spread, and regresses their polarization from the pro-tumor M2 phenotype to the tumor growth inhibitory M1 phenotype.27,30
Recently, a synergistic relationship between regorafenib and natural killer (NK) cells has been reported. 36 Binding between NKG2D receptors on the surface of NK cells and NKG2DL expressed in tumor cells leads to NK cells activation that eliminates tumor cells. However, tumor cells utilize various mechanisms to evade NKG2D/NKG2DL receptor-mediated immune clearance; NKG2D-expressing chimeric antigen receptor T (CAR-T)-cell therapy has exhibited robust antitumor efficacy in preclinical HCC models.37–39
Tai and colleagues demonstrated that regorafenib induces inhibition of the signal transducer and activator of transcription 3 (STAT3) signaling pathway. This results in increased cytolytic activity of NK cells through upregulation of the NKG2D ligand and the recognition of HCC cells by NK cells, and, ultimately, apoptosis of HCC cells. 40
Overall, a growing body of evidence has demonstrated that the modulation and promotion of anti-tumor immunity by regorafenib is seen through its effects on various components of the TME (Figure 3). 22
Finally, long-term therapy with regorafenib may also reduce angiogenesis and be beneficial for portal hypertension; acute administration improves portal hemodynamics, indicating that it may be particularly beneficial for patients with portal hypertension and preserved liver function. 41
As such, regorafenib’s broad spectrum of kinase inhibition, coupled with its immunomodulatory effects, may explain its established and emerging clinical activity in various tumor types. This has supported a rationale for supporting clinical trials to investigate the development of a combination strategy with immune checkpoint inhibitors.42–44
Regorafenib is metabolized by the enzymes UDP-glucuronosyltransferase 1-9 (UGT1A9) and cytochrome P450 3A4 (CYP3A4) into two active metabolites, demethylated N-oxide (M-5) and N-oxide (M-2) (Figure 1, Panel B).29,45
CYP enzymes may be inhibited or induced by co-administration of agents that interact with the same enzymes. Co-administration of regorafenib with a strong CYP3A4 inhibitor may extend the regorafenib serum area under the curve (AUC). This could result in a potential increase in the drug toxicity. Conversely, co-administration of regorafenib with a significant CYP3A4 inducer would lead to a decrease of regorafenib in serum AUC levels and a potential impairment of efficacy. The M-5 and M-2 metabolites of regorafenib also affect CYP isoenzymes as they have been shown to inhibit cytochrome P450 family 2 subfamily C member 9 (CYP2C9), the enzyme responsible for metabolizing warfarin), cytochrome P450 2B6 (CYP2B6), CYP3A4, and cytochrome P450 family 2 subfamily C member 8 (CYP2C8). 46
Table 2 lists the agents that potentially interact with regorafenib and the effects of these interactions. In view of potential drug interactions, drug compatibility should be verified in all patients before starting treatment with regorafenib.
Major drug interactions with regorafenib.

Inducers of CYP3A4 may decrease exposure to regorafenib and exposure to M-2 and M-5 metabolites may increase.
Inhibitors of CYP3A4 may increase exposure to regorafenib and exposure to M-2 and M-5 metabolites may decrease.
Regorafenib inhibits CYP2C9; concomitant administration of drugs that are CYP2C9 substrates may result in increased exposure of that drug.
Regorafenib is a UGT1A1 inhibitor: concomitant use with irinotecan may result in increased irinotecan exposure.
CYP2C9, cytochrome P450 family 2 subfamily C member 9; CYP3A4, cytochrome P450 3A4; M-2, demethylated N-oxide; M-5, N-oxide; UGT1A1, UDP-glucuronosyltransferase 1A1.
The efficacy and safety of regorafenib in preclinical studies
In xenograft models, regorafenib was found to inhibit tumor growth of extracellular signal-regulated kinase phosphorylation, which could be shown by an expressive reduction in the tumor areas microvessel density.29,47,48
A phase II study involving 36 HCC patients showed both acceptable tolerability and a proven antitumor activity, with a median OS of 13.8 months and a median time to progression (TTP) of 4.3 months. 49
These results led to the RESORCE study design, a phase III placebo-controlled trial, which included patients who had progressed on sorafenib but tolerated ⩾400 mg/d for at least 20 of the last 28 days of treatment. 15 The last dose of sorafenib had to have been received within the last ten weeks before randomization. A 2-week wash-out from the last dose of sorafenib was mandatory before starting regorafenib, while exclusion criteria included discontinuation of sorafenib due to toxicity.
The trial was carried out in 152 centers across 21 different countries and four continents. Participants were assigned randomly (2:1) to 160 mg oral regorafenib or placebo once daily for 3 weeks, followed by 1-week off. A total of 4 weeks constituted one full treatment cycle. All patients received best supportive care.
The study’s primary endpoint was OS time (time from randomization to death), analyzed by intention to treat. Secondary endpoints were TTP, progression-free survival (PFS), objective response rate (ORR) [complete response (CR) or partial response (PR)], and disease control rate (CR, PR, or stable disease maintained for ⩾6 weeks) as estimated by the investigators by applying mRECIST and RECIST 1.1 criteria.
Patients were stratified by geographical region (Asia versus rest of the world), the presence of macrovascular invasion (yes versus no), the presence of extra-hepatic disease (yes versus no), α-fetoprotein concentration (<400 versus >400 ng/ml), and Eastern Cooperative Oncology Group performance status (0 versus 1). After screening 843 patients, 573 patients were enrolled and randomized (379 to the regorafenib arm and 194 to the placebo arm).
A total of 216 patients were from Asia. Before starting regorafenib, the median treatment duration with sorafenib was 7.8 months [interquartile range (IQR) 4.2–14.5] in the regorafenib arm and 7.8 months (IQR: 4.4–14.7) in the placebo arm. The median treatment time on regorafenib in this study was 3.6 months (IQR: 1.6–7.6) and 1.9 months (IQR: 1.4–3.9) on placebo.
The median daily dose of regorafenib was 144.1 mg. Importantly, regorafenib therapy resulted in a significantly higher OS with a median OS of 10.6 months (95% CI: 9.1–12.1) compared with 7.8 months (6.3–8.8) under placebo (HR: 0.63; 95% CI: 0.50–0.79; p < 0.0001), with a 37% reduction in the risk of death. The median PFS was also significantly improved in the regorafenib group at 3.1 months (95% CI: 2.8–4.2) compared to 1.5 (1.4–1.6) months in the placebo group. This amounted to a 54% reduction in the risk of progression or death (HR: 0.46; 95% CI: 0.37–0.56; p < 0.0001).
The median TTP in the regorafenib arm was 3.2 months (2.9–4.2 95% CI) with regorafenib compared with 1.5 (1.4–1.6) months in the placebo arm (HR: 0.44; 95% CI: 0.36–0.55; p < 0.0001). A total of 11% of the regorafenib-treated patients compared with 4% in the placebo arm achieved an objective response (p = 0.0047). Two patients (1%) in the regorafenib arm versus zero patients in the placebo arm achieved a CR.
The alpha-fetoprotein (AFP) response rate, defined as a ⩾20% decrease in AFP from baseline to the start of cycle 3, was higher in patients treated with regorafenib than those receiving placebo.
The toxicity profile of regorafenib is similar to that of other TKIs, especially sorafenib. In early phase studies, dose-limiting toxicities included bone marrow suppression and gastrointestinal toxicities.29,45,47 In a phase II study in HCC patients, 58% of patients experienced an adverse event (AE) of grade 3 or higher. 49 These included fatigue (17%), hand–foot skin reaction (HFSR) (14%), and diarrhea (6%). A total of 19% of patients discontinued treatment due to AEs that the investigator judged to be treatment-related.
In the phase III RESORCE trial, AEs were recorded in all regorafenib-treated patients (100%) and 179 of the 193 placebo-receiving patients (93%). 15 The most frequent clinically significant grade 3 or 4 treatment-emergent adverse events (TEAEs) were hypertension (15%), HFSR (13%), fatigue (9%), and diarrhea (3%). A total of 10% of patients experienced regorafenib-related serious AEs, and seven deaths (2%) were attributed to the study drug versus 2% in the placebo group.
The only drug-related deaths due to liver failure were observed in the placebo group. A total of 6% of patients in the regorafenib-treatment arm experienced grade 3 or higher TE bleeding events compared with 8% in the placebo arm.
A total of 255 (68%) of the 374 patients in the regorafenib arm underwent dose interruptions or reductions due to AEs, compared with 60 (31%) of the 193 placebo-receiving patients.
Overall, regorafenib was well tolerated. Drug-related AEs led to discontinuations or dose reductions in 202 (54%) patients in the regorafenib arm and 20 (10%) in the placebo arm; discontinuation owing to a treatment-related AEs were relatively low, at 39 (10%) in the regorafenib arm, compared with 7 (4%) in the placebo arm.
The most common AEs causing treatment discontinuation most frequently observed with regorafenib were: (i) increased aspartate transaminase (AST) [8 (2%) of 374 patients versus 3 (2%) in the placebo group], (ii) HFSR [7 (2%) versus none], and (iii) increased alanine transaminase (ALT) [4 (1%) versus none].
Additional sub-analyses of the RESORCE study were performed and showed that: (I) a longer survival follow-up, almost 1 year after the pivotal analysis, confirmed the initial primary OS result; 50 (II) comparing tumor response and progression in the RESORCE study using mRECIST and RECIST 1.1 criteria, although a slightly higher response rate was observed using the mRECIST ones, PFS, TTP, and disease control rates were not different when assessed by investigators using mRECIST or RECIST 1.1 criteria;51,52 I(II) an exploratory analysis, aimed at validating the concept of progression profile in a global cohort of patients previously treated with sorafenib and assessing the impact of regorafenib on survival by looking at prior progression, showed that regorafenib provides an OS benefit regardless of progression pattern; 52 (IV) patients who develop HFSR under regorafenib tended to have a better OS [median OS, 14.1 months (95% CI 11.7, 16.5) versus 6.6 (5.0, 8.5)], as previously demonstrated for sorafenib.53–55
Finally, an adjunctive exploratory analysis of the RESORCE trial reported that patients who received the sorafenib-regorafenib sequence achieved a median OS of 26 months, a result never previously demonstrated. 56 Analysis showed that treatment with regorafenib yielded a clinical benefit regardless of the last dose of sorafenib or the TTP on sorafenib.
Regorafenib treatment in real-life clinical practice
Since the approval of regorafenib in 2017, clinical practice studies have provided results, although still limited, on regorafenib’s safety and efficacy profile in real-life experience.57–60
REFINE [ClinicalTrials.gov identifier: NCT03289273] is an ongoing observational study that recruited patients with HCC for whom the decision to treat with regorafenib was taken by the treating physician before enrollment, according to the label approved by the local health authority. 57
The interim analysis results performed after the first 500 enrolled patients were presented during the 2020 International Liver Cancer Association (ILCA). 58
REFINE has a larger patient population than RESORCE, reflecting less stringent inclusion criteria than real-world studies. Most patients (67%) had Child–Pugh class A liver function; 11% and 1% had Child–Pugh class B and C liver function, respectively (Child–Pugh score was missing or not assessable in 21% of patients).
The proportions of patients with Eastern Cooperative Oncology Group performance status (ECOG PS) 0, 1, and 2-4 were 42%, 40%, and 5%, respectively (ECOG PS was missing or not evaluable in 13% of patients). Most patients (98%; n = 490) received previous systemic therapy; 97% (n = 482) had previously received sorafenib. Regorafenib was a second-line treatment in 81% of patients (n = 403), third-line or higher in 17% (n = 87), and first-line in 2% (n = 8).
Of the 403 patients who received regorafenib second-line treatment, 398 (99%) had received sorafenib first. Among all treated patients (n = 498), 57% (n = 286) started regorafenib at a daily dose of 160 mg, 13% (n = 63) at 120 mg, 28% (n = 141) at 80 mg, and 2% (n = 8) at 40 mg.
In the 482 patients who had received sorafenib in any prior line of therapy, the median duration of prior sorafenib was 4.8 months (IQR: 2.5–9. 6). Two hundred and sixteen (45%) patients had a last daily dose of sorafenib of 800 mg, 8% of patients (n = 40) had developed a side effect that led to discontinuation of sorafenib (defined as sorafenib intolerant patients) and, at study entry, the percentages of patients with Child–Pugh class A, B, and C liver disease were 67%, 12%, and 1%, respectively.
Among all patients treated with regorafenib (n = 498), the most frequent TEAEs (any grade) were HFSR (30%), diarrhea (21%), fatigue (16%), and decreased appetite (14%). In patients intolerant to sorafenib, the most frequent TEAEs (any grade) with regorafenib were diarrhea, HFSR, abdominal pain, and decreased appetite.
The investigators assessed OS by Child–Pugh class and albumin-bilirubin (ALBI) grade at study entry in patients who had previously received sorafenib. The median OS was 16.0 months among the Child–Pugh class A group (95% CI, 13.1–18.8) versus 8.0 months among the Child–Pugh class B group (95% CI, 5.2-not evaluable). The median OS among those with ALBI grade 1, 2, and 3 was 19.6 months (95% CI, 14.8–19.6), 10.5 (95% CI, 8.7–16.0), and 3.1 months (95% CI, 1.6–8.7), respectively.
Regorafenib confirmed the survival benefit regardless of the disease progression rate during previous treatment with sorafenib or since the last dose of sorafenib also in a retrospective safety and efficacy study in Korean patients where data were consistent with those from the RESORCE study. 59
In a subsequent multicenter, retrospective analysis of 440 patients who had previously received sorafenib and were treated with regorafenib as a second (69.3%), third (26.1%), and fourth to seventh (4.5%) line of therapy at nine tertiary referral hospitals in Korea, actual clinical outcomes were consistent with the RESORCE study results, and regorafenib-related HFSR was significantly associated with improved OS. 60
Interestingly, intracavernosal injections (ICIs) were administered in 115 patients (26.1%) before regorafenib; there were no differences in PFS and OS with regorafenib related to previous use of ICIs.
A clinically relevant aspect arising from some clinical practice studies is the importance of the patient’s physical status and residual liver function after first-line failure. These parameters affect the rate of patients eligible for switching to second-line agents after radiological progression with first-line treatment with sorafenib.
A Canadian study characterized the sequential therapies received by HCC patients after sorafenib. It also determined the rate of patients eligible for new therapies if strict eligibility criteria (SEC; as defined in the respective studies) were used, compared with more liberal modified eligibility criteria (MEC, including Child–Pugh-B7 and ECOG 2). 61
Overall, 730 patients were identified and 172 (23.6%) received subsequent treatment (regorafenib, Cabozantinib, or ramucirumab). Patients who received a subsequent treatment had a significantly longer OS than patients who did not have access to it (12.1 versus 3.3 months; p < 0.001). Using SEC, only 13.1% of patients would be eligible for second-line treatment. Extending eligibility to patients meeting MEC, however, increased the eligibility rate to 31.7%.
The highest ineligibility for regorafenib was determined by study-specific criteria, including sorafenib intolerance (28%).
This study showed that only a restricted proportion of HCC patients in the real world would be eligible for Cabozantinib, regorafenib, or ramucirumab if the SEC of clinical trials were followed; whereas, more than twice as many would be eligible if the MEC were followed. Patients who received subsequent treatment had a better OS, regardless of whether they met SEC or MEC.
A small Japanese retrospective study reported that only about 30% of patients refractory to first-line sorafenib therapy were eligible for second-line regorafenib treatment in clinical practice. 62 The main reasons patients could not be treated with regorafenib were sorafenib intolerance and liver function deterioration.
This and other real-life studies emphasize that in order to extend the prognosis with the use of effective second-line therapies, it is essential to preserve liver function before and during both previous transarterial and first-line therapies. 63
A preserved liver function and ECOG performance status during treatment with sorafenib accounted for the subsequent treatment’s efficacy and improved outcome.64,65
This is corroborated by the finding that the novel biomarker of liver reserve function, ALBI grade, was able to select regorafenib candidates successfully. A median OS of 15.6 months was achieved in the identified cohort compared with 6.8 months for non-candidates. 66
Yukimoto et al. reported that an ALBI grade of −2.53 at the time of sorafenib initiation was helpful as a threshold value for the prediction of regorafenib eligibility following the failure of sorafenib. 67
Takada et al. confirmed in a recent study that a more careful estimation of liver function has emerged as an essential prerequisite in this setting. 68 They showed that at the time of first-line sorafenib failure, the criteria for inclusion in the RESORCE study were not just the baseline ALBI score (−2.33; OR 2.5, p = 0.01) but also the degree of change in liver function after four weeks of treatment with sorafenib (<0.255; OR 4.9, p < 0.001).
Similarly, Moriguchi et al. demonstrated that the ALBI grade at the beginning of sorafenib therapy is a significant factor correlated with maintenance of Child–Pugh A class and ECOG-PS ⩽1 upon discontinuation of sorafenib. It is a good indicator of the possibility of introducing second-line therapy after sorafenib. 69
Accordingly, a recent small retrospective study suggests that regorafenib’s clinical outcomes and higher frequency of serious adverse events would discourage its use in Child–Pugh B patients with grade 3 ALBI. 70
As there are still no proven biomarkers in clinical practice to guide systemic therapy, a Japanese study aimed to evaluate relative dose intensity (RDI), defined as the ratio of administered dose to planned dose, and the association between RDI and OS in patients with unresectable HCC. 71
Patients with first-month RDI ⩾ 50% were shown to have significantly better OS and PFS than those with first-month RDI < 50% [HR 0.19 (CI 0.08–0.48), p = 0.0004 and HR 0.2 (CI 0. 08–0.52) p = 0.0008], and a first-month RDI ⩾ 50% [HR 0.18 (CI 0.06–0.55) p = 0.002] and a HSFR [HR 0.03 (CI 0.008–0.16) p < 0.0001] were independently correlated with OS.
Therefore, sorafenib-regorafenib sequential treatment was effective and well-tolerated in Japanese patients with unresectable HCC. A first-month RDI of ⩾50% regorafenib has shown clinical relevance and, if confirmed in more extensive studies, could be a valuable tool to guide second-line therapy.
Finally, regorafenib has also been shown to be effective in sorafenib-tolerant patients with recurrent HCC after liver transplantation who develop progression, in a retrospective, multicenter, international study reporting a median OS of 12.9 months after regorafenib initiation and 38.4 months (18.5–58.4 95% CI) for sorafenib-regorafenib sequential treatment. The AEs reported in the study were not only similar to those reported in the registrative study, but were also comparable to those that emerged in the same patients during the previous sorafenib treatment. 72
Predictive and/or prognostic markers of regorafenib in HCC
Following the progress of new effective systemic therapies for HCC, the current challenge is selecting patients to determine the appropriate treatment choice.
The identification of useful predictive markers for clinical outcomes associated with regorafenib treatment is crucial; however, to date, no established biomarkers have been identified.
In the absence of clinical/biological predictors to identify potentially responsive patients, a retrospective biomarker analysis was conducted on patients enrolled in the RESORCE trial to identify biomarkers potentially predictive of benefit for regorafenib in HCC. 73
Plasma and tumor samples from RESORCE study participants were assessed in 567 patients (374 regorafenib and 193 placebo arm) to identify genetic, microRNA (miRNA), and protein biomarkers correlated with response to regorafenib.
Notably, nine plasma miRNAs (MIR30A, MIR122, MIR125B, MIR200A, MIR374B, MIR15B, MIR107, MIR320, and MIR645) were significantly associated to increased OS time under regorafenib. Decreased levels of five proteins [angiopoietin 1 (ANG-1), cystatin B, transforming growth factor-beta 1 latency-associated peptide (LAP TGF-b1), oxidized low-density lipoprotein receptor 1 (LOX-1), and C-C motif chemokine ligand 3 (MIP-1a)] were identified as predictors of the benefit of regorafenib treatment.
This is currently the unique study that provides a potential biomarker-guided strategy for identifying patients conceivably responsive to regorafenib, but it still needs validation in further studies.
It has been suggested that TIE2 is a potential circulating biomarker of tumor vascular response for VEGF inhibitor, assuming that TIE2 originates from the tumor blood vessels. 74
As the oncological use of anti-angiogenic VEGF inhibitors has been limited by the absence of informative biomarkers, circulating TIE2 could be a candidate tumor vascular response biomarker for VEGF inhibitors.
Interestingly, during regorafenib treatment, a dynamic change of plasma angiogenic components has been described: low baseline levels of ANG-2 and TIE2 appear to be linked to a better prognosis. Early modulation of ANG-2 levels may be predictive of response to regorafenib in patients with metastatic colorectal cancer.
Such results would support an exploratory study to verify this prognostic correlation in HCC patients. 75
The combination of regorafenib with other treatments
In recent years, encouraging data that would promote the combination of regorafenib’s antiangiogenetic effects with ICIs to optimize and enhance the two individual therapies’ response rates has been reported.21,22,76,77
Since the TME, a key determinant of tumor growth and metastasis, is characterized by various counterparts, including immune and non-immune cell populations and non-cellular components, the combination of current TKIs with immunotherapy has been investigated to exploit maximal therapeutic benefit.21,78,79
Regorafenib within the sub-micromolar range induced M1 macrophage polarization and enhanced CD8+ T cell proliferation and activation (Figure 2). Also, in vivo studies using regorafenib at low-dose (3–5 mg/kg/day, representing approximately 50% of the recommended single-agent dosage in the clinic) showed synergistic antitumor efficacy with anti-program cell death-1 (PD-1) therapy.77,79
Recognizing the optimal immunomodulatory-effects of targeted-agents is essential for the development of combination immunotherapy. This helps to improve the therapeutic index and to tailor the use of targeted drugs to their biologically active and clinically significant dosage. 21
In a preclinical study, it has been reported that in a combination treatment strategy with anti-PD-1 antibody, regorafenib can significantly intensify PD-1 blockade effects in a dose-dependent manner. 42
The benefit was the result of the activity of the two agents on both normalization of the HCC vasculature and stimulation of anti-tumor immunity. The combination treatment inhibited STAT3 activity and raised the expression of the C-X-C motif chemokine ligand 10 (CXCL10). This extended both tumor penetration and the survival of activated CD8 T cells.
This concept is clinically relevant for the future design of combination treatment strategies in HCC patients.
The potential synergistic antitumor efficacy of regorafenib with anti-PD-1 therapy has also been shown in a study of an orthotopic HCC model. This has proved that regorafenib may modulate macrophage polarization, increase T cell activation, and, thereby, enhance the efficacy of anti-PD-1 therapy. 80 Therefore, the optimization of regorafenib dosage for the rational design of combination therapy regimen may improve the therapeutic index in the clinic.
In a recent phase open-label, dose-escalation phase Ib study, another TKIs/ICI combination treatment based on regorafenib plus pembrolizumab, an anti-PD-1 monoclonal antibody, was investigated in patients with advanced HCC who received no previous systemic treatment [ClinicalTrials.gov identifier: NCT03347292]. 81
In the first cohort, patients underwent regorafenib 120 mg/day orally for three weeks on/1 week off with pembrolizumab 200 mg intravenously for 3 weeks. Thereafter, the regorafenib dose could be increased (160 mg) or lowered (80 mg), according to the modified toxicity probability interval design, while the dosage of pembrolizumab was steady. The primary endpoints were tolerability and safety. The secondary aims were to determine the maximum tolerated dose (MTD) and recommended phase II dose and to evaluate anti-tumor efficacy.
Twenty-nine patients received regorafenib 120 mg dosage. The median age was 65 years (range 32–81); 41% and 55% of patients had BCLC stage B and C respectively, while 100% were Child–Pugh A class; ECOG status 1/0 was 28%/72%. Dose-limiting toxicities were recorded in 4/18 evaluable patients: grade 3 raised AST/ALT with grade 2 raised bilirubin (n = 2); grade 3 rash (n = 2). The MTD of regorafenib in the combined treatment was 120 mg.
There were no grade 5 TEAEs. Dose modifications (interruption and/or dose reduction) of regorafenib/pembrolizumab for drug related TEAEs were reported in 59%/31% of patients.
Of 23 assessable patients, 7 (30%) exhibited a partial response while 14 (61%) showed stable disease (according to RECIST v1.1). One additional patient had a partial response (according to mRECIST). As a result, the combined treatment with regorafenib plus pembrolizumab as first-line therapy of advanced HCC showed encouraging antitumor activity and safety profiles.
Enrollment has been sustained and is ongoing at regorafenib 120 mg dose.
Nivolumab is a human immunoglobulin G4 (IgG4) monoclonal antibody to the PD-1 receptor, which blocks the interaction with PD-ligand (PD-L)1/PD-L2, thereby resuming T-cell-mediated antitumor effects. It was approved in 2017 for the second-line treatment of HCC patients who have been previously treated with sorafenib. 18
A phase I/IIa trial [ClinicalTrials.gov identifier: NCT04170556] is ongoing and is aimed to assess the effects of nivolumab and regorafenib, while recognizing the potential impact of the interaction of drugs and enhanced severity and/or frequency of AEs. 82
Therefore, regorafenib will be given as monotherapy during the first two cycles (each cycle is 3 weeks on plus 1 week off) of treatment to enhance T cell trafficking and infiltration into the tumor bed to increase the benefits of anti-PD-PD-L1.
Tislelizumab is a humanized monoclonal antibody directed against PD-1, currently tested for hematological cancers and advanced solid tumors. 83
An ongoing phase II study [ClinicalTrials.gov identifier: NCT04183088] will investigate the efficacy and safety of the combined tislelizumab with regorafenib as first-line treatment for advanced HCC. 84
This trial consists of 2 parts. Part 1 consists of a single-arm study, and eligible patients will be assigned tislelizumab 200 mg intravenously on day 1 every 3 weeks, plus regorafenib (80 mg/d). Part 2 is a randomized study. Subjects will be 1:1 randomized to two treatment arms: (1) tislelizumab and regorafenib combined treatment used in part 1, versus (2) regorafenib and placebo. For patients in group 2, when imaging assessment shows SD or PD, according to RECIST v1.1 criterion, the therapeutic strategy will be shifted to tislelizumab + regorafenib combination schedule.
Finally, a multicenter, open-labeled prospective phase Ib trial [ClinicalTrials.gov identifier: NCT03475953] investigating three dosage levels of regorafenib combined with avelumab, a human IgG1 monoclonal antibody that targets PD-L1, in both advanced and metastatic solid tumors (including HCC), is currently being recruiting. 85
Ongoing clinical trials based on combined regorafenib/ICI agents are reported in Table 3.
Ongoing clinical trials with regorafenib-based combination treatments (www.clinicaltrials.gov).
Patients progressing Under first-line Sorafenib.
RP2D, recommended phase II dose.
cTACE, conventional transarterial chemoembolization; DEB-TACE, drug-eluting bead transarterial chemoembolization; IL, interleukin; IV, intravenous; PD-1/PD-L1, programmed cell death protein-1/ligand; RP2D, recommended phase II dose; TACE, transarterial chemoembolization.
In addition to immunotherapy, other combination treatments of regorafenib with agents acting on parallel and complementary pathogenic pathways have also been reported in preclinical cancer models. 22
Annexin A3 (ANXA3) is recognized to have a key role in enhancing tumor aggressiveness, preventing apoptosis, and promoting pro-survival autophagy in sorafenib-resistant HCC cells.
Tong et al. demonstrated in in vivo models of sorafenib unresponsive HCC that co-administration of regorafenib and an anti-ANXA-3 monoclonal antibody can potentiate apoptotic induction by abrogating autophagy. 86 Likewise, navitoclax, a specific inhibitor of the anti-apoptotic proteins B-cell lymphoma 2 (Bcl-2) and B-cell lymphoma-extra large (Bcl-xL), enhanced the regorafenib sensitivity of Hep3B and HepG2 cells, as evidenced by enhanced apoptotic features. 87
The potential benefit of regorafenib has also been tested in combination treatment with transarterial chemoembolization (TACE).
Regorafenib-loaded polylactide-co-glycolic acid (PLGA) microspheres for improvement of TACE therapeutic effects, which can sustainably deliver regorafenib to limit proangiogenic responses in liver tumors after TACE, has been recently developed. 88 The fabricated regorafenib microspheres provided sustained drug release for more than 30 d in vitro and in vivo after TACE. The study demonstrated that the new regorafenib microspheres, as a form of local drug delivery combined with TACE, may enhance the therapeutic potency of TACE for the treatment of HCC, and has promising clinical implications in future.
Discussion
Recent studies have started to decode the complexity of the HCC immune microenvironment, such as the function and subsets of different immune cells in the liver, including T and B cells, macrophages, neutrophils, NK cells, dendritic cells, myeloid-derived suppressor cells, cancer-associated fibroblasts, and the active interplay between immune cells and the cancer ecosystem in promoting angiogenesis. 89
These data establish a strong rationale supporting a therapeutic strategy that simultaneously targets the main pathogenic pathways that favor tumor proliferation, spread, and neoangiogenesis as well as the immune mechanisms that allow tumor cells to elude immune suppression. 43
This combined approach is highly likely to lead to an enhancement of anti-tumor therapies with maximized response rates and the chance of obtaining not only disease stabilization but also tumor mass shrinkage with a higher objective response rate.
The combined use of atezolizumab with bevacizumab in first-line unresectable HCC has newly shown a superior benefit over sorafenib in a recently published phase III trial. This has confirmed that by targeting simultaneously the pathogenic pathways that support tumor growth, spread, and neoangiogenesis on the one hand, and immunosuppression and tumor-induced immune evasion on the other, the efficacy of anti-tumor treatments can be significantly augmented. 14
However, these therapies are utilized in clinical practice with not as strict inclusion criteria as the respective phase III studies.20,61,70 For instance, in the prospective observational study (REFINE) of regorafenib in HCC patients, 11% of treated patients had Child–Pugh B liver function, and 28% of total patients were initiated on 80 mg of regorafenib rather than the standard 160 mg dose. These dose modifications to attenuate TKI-related AEs without affecting efficacy have been prospectively analyzed in metastatic colorectal cancer, where a dose-escalation approach to regorafenib showed favorable AEs and comparable therapeutic efficacy to the entire dose. 90
Equally, sorafenib at 200 mg proved more tolerable than the 400 mg dose with comparable efficacy in large retrospective studies.91,92
A recent meta-analysis, based mainly on real-world studies examining regorafenib as second-line therapy after sorafenib failure, confirm the promising favorable outcomes observed with the RESORCE trial. The also confirm that regorafenib provides both a valid and safe treatment strategy in patients with intermediate/advanced HCC who exhibit disease progression on sorafenib. 93
In the near future, new clinical trials for HCC patients should be aimed at investigating the potential benefit and synergistic effects of regorafenib with ICIs.
A very relevant issue that remains is the treatment of patients with suboptimal liver function, who have historically been excluded from registrative studies. In order to design clinical trials that also include more fragile patients, it would be appropriate to investigate the tolerability of regorafenib in patient categories not included in the registrative study through real-world clinical practice studies. In this regard, it would be crucial to test the safety and efficacy of regorafenib in patients with an imperfect liver function (Child–Pugh B7) and currently with limited systemic treatment options, following the clinical practice experience of sorafenib therapy.63,94
A key step in the development of improved systemic treatment strategies for HCC is the identification of clinical-biological markers predictive of response that would be critical for optimally selecting patients who may benefit from regorafenib and other therapies.73,74
Novel combination treatment approaches of regorafenib with other ICIs provide an exciting opportunity for continued research. A breakthrough for patients with unresectable HCC is expected in the near future. Therefore, it will be crucial to have a thorough knowledge of the pharmacological characteristics of each drug and the most appropriate management of possible side effects to achieve maximum therapeutic benefit.
Footnotes
Author contributions
AF and FT reviewed the literature and drafted the manuscript; AF, FP, AG, and MR analyzed and critically interpreted literature data; FP, FT, AF, SM, MR, LI, FB and VS contributed to manuscript drafting; all authors were involved in acquisition of data; all authors critically reviewed the manuscript and approved the final version of this manuscript.
Conflict of interest statement
FP: (honoraria for speaker bureau, consultancy or advisory board) Alkermes, Astrazeneca, Bayer, Bracco, BMS, EISAI, GE, MSD, IPSEN, La Force Guerbet, Roche, Siemens Healthneers, Tiziana Life Sciences. Research contract with ESAOTE.
FT: consultant for Bayer, Speaker bureau honoraria from MSD, Grant from Ipsen.
AG, SM, AF, MR and FB have no conflict of interest to declare.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.