
Abstract
Background:
The effect of low-frequency functional variation on anti-tumor necrosis factor alpha (TNF) response in Crohn’s disease (CD) patients remains unexplored. The objective of this study was to investigate the impact of functional rare variants in clinical response to anti-TNF therapy in CD.
Methods:
CD anti-TNF naïve patients starting anti-TNF treatment due to active disease [Crohn’s Disease Activity Index (CDAI > 150)] were included. The whole genome was sequenced using the Illumina Hiseq4000 platform. Clinical response was defined as a CDAI score <150 at week 14 of anti-TNF treatment. Low-frequency variants were annotated and classified according to their damaging potential. The whole genome of CD patients was screened to identify homozygous loss-of-function (LoF) variants. The TNF signaling pathway was tested for overabundance of damaging variants using the SKAT-O method. Functional implication of the associated rare variation was evaluated using cell-type epigenetic enrichment analyses.
Results:
A total of 41 consecutive CD patients were included; 3250 functional rare variants were identified (2682 damaging and 568 LoF variants). Two homozygous LoF mutations were found in HLA-B and HLA-DRB1 genes associated with lack of response and remission, respectively. Genome-wide LoF variants were enriched in epigenetic marks specific for the gastrointestinal tissue (colon, p = 4.11e–4; duodenum, p = 0.011). The burden of damaging variation in the TNF signaling pathway was associated with response to anti-TNF therapy (p = 0.016); damaging variants were enriched in epigenetic marks from CD8+ (p = 6.01e–4) and CD4+ (p = 0.032) T cells.
Conclusions:
Functional rare variants are involved in the response to anti-TNF therapy in CD. Cell-type enrichment analysis suggests that the gut mucosa and CD8+ T cells are the main mediators of this response.
Introduction
Anti-tumor necrosis factor alpha (TNF) drugs have been shown to be effective for induction and maintenance of remission in Crohn’s disease (CD); however, up to 30% of patients do not respond to these drugs.1–3 In 2014, the use of vedolizumab, which targets α4β7 integrins, was approved for CD. In 2016, ustekinumab, directed against the p40 subunit shared by interleukins 12 and 23, was approved in patients with CD, while the JAK-inhibitor tofacitinib has just been approved by the European Agency of Medicines for ulcerative colitis (UC) patients, hence increasing the therapeutic arsenal against inflammatory bowel diseases (IBD). All these drugs have high cost, so they pose a great economic burden on health systems. At present, the medical community does not have reliable criteria for selecting which patient will benefit from any of the above-mentioned drugs. Thus, the variables (epidemiological, clinical, analytical, etc.) commonly used to predict patient response to biologic therapy have shown low utility.
Therefore, it is a priority to identify mechanisms involved in the response to each drug, to suggest more rational use of resources and for personalized medicine. Thus, achieving that goal will allow us to indicate the most appropriate treatment to each individual, hence avoiding administering drugs to patients who will not respond (an inadequate use of resources and an unjustified risk of adverse effects).
There is compelling evidence that the clinical response to anti-TNF therapy has a genetic component. 4 To date, several candidate-gene studies have investigated the contribution of common variation to the heterogeneity of anti-TNF response in CD.5–9 However, the few associations identified so far have not been consistently replicated. 10 The study of the genetic basis of the clinical response to anti-TNF therapy at the genome-wide scale represents a new opportunity to identify genetic factors underlying the anti-TNF response in CD.
The advent of next generation sequencing (NGS) technologies like whole genome sequencing (WGS) is enabling the analysis and identification of low-frequency variation associated with complex traits,11,12 including the response to anti-TNF agents. 13 In particular, rare variants with a high functional impact can display much larger effects on complex phenotypes than common genetic variants. 14 From a clinical perspective, loss-of-function (LoF) variants are one of the most interesting forms of rare functional genetic variation. 15 Accordingly, in the pharmacogenomics field, the analysis of LoF variants is starting to provide new insights into the genetic basis underlying the clinical response to therapies in complex diseases. The analysis of low-frequency variation could therefore shed new light on the biological mechanisms that are relevant for anti-TNF response in CD.
In the present study, we sequenced the whole genome of CD patients treated with anti-TNF therapy. At the genome-wide level, we identified LoF variants affecting critical genes for the immune response. Specific analysis of the TNF signaling pathway has also shown the significant burden of damaging variation associated with treatment response. These findings support the importance of functional rare variation for the anti-TNF response in CD, and could help to select those patients who will benefit most from anti-TNF treatment.
Methods
Patients
A total of 41 patients diagnosed with CD based on European Crohn’s and Colitis Organization criteria that started anti-TNF treatment were included. 16 Patients came from the Predicrohn study, which is a multicenter study aiming to optimize treatment with anti-TNF drugs. 17 The trial included 150 CD patients naïve to anti-TNF treatment; only patients with active disease, based on the Crohn’s Disease Activity Index (CDAI) at baseline, were eligible for the genetic approach. All patients were naïve to anti-TNF therapy and received 160/80 mg adalimumab at weeks 0 and 2, and 40 mg every other week thereafter, or infliximab 5 mg/kg at weeks 0, 2, 6, and 14, and every 8 weeks thereafter.
Study design
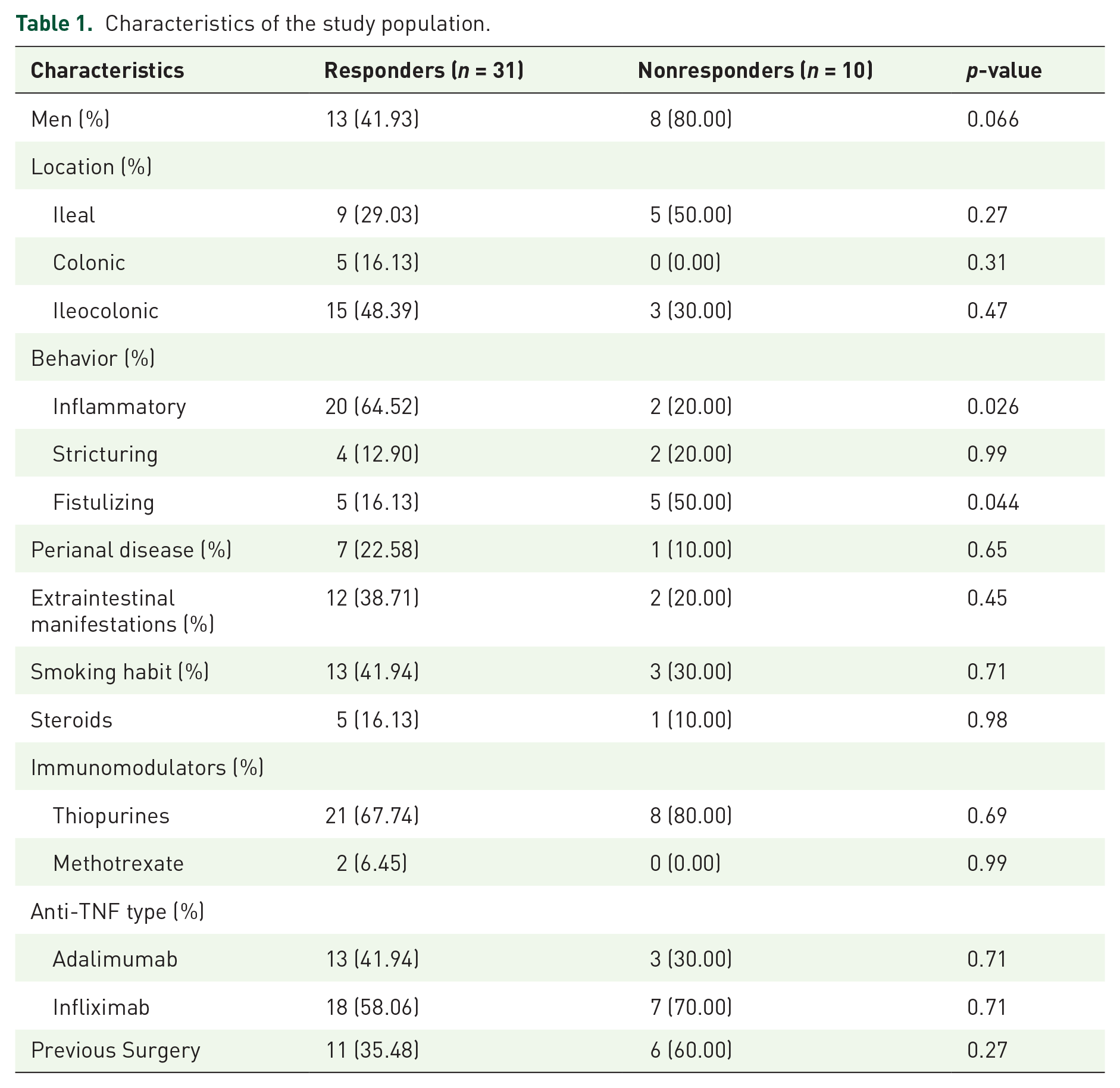
We conducted a prospective, multicenter cohort study. Treatment (infliximab or adalimumab) was not assigned randomly but based on the clinician’s decision. Clinical response was evaluated at week 14 by measuring the change in the CDAI score. Remission was defined as CDAI⩽150 at week 14, and short-term partial response as a decrease in the CDAI score of at least 70 points at week 14, without achieving remission. Patients that achieved remission, and those with partial response, were considered responders. Patients not meeting those criteria were considered nonresponders. Concomitant treatments were also recorded. Blood samples were obtained at baseline. The distribution of the categorical variables between responders and nonresponders to anti-TNF therapy was compared using the Fisher’s exact test (Table 1).
Characteristics of the study population.
The study was approved by the ethical review board of all participating centers. All patients signed an informed written consent to participate.
WGS of the study population
WGS of the 41 CD patients included in the present study was performed using the Illumina Hiseq4000 platform at Centre Nacional d’Anàlisi Genòmica (CNAG, Barcelona, Spain). All DNA samples were sequenced to an average coverage >20× using 150 bp paired-end reads. For each sample, we obtained >360 million reads. Duplicate reads were subsequently excluded from the analysis. The remaining reads were aligned to the GRCh38/hg38 assembly of the human reference genome using the Burrows Wheeler algorithm. Variant calling was performed according to the best practices outlined in the Genome Analysis ToolKit (GATK). 18
In the quality control (QC) analysis, genetic variants with a genotype quality ⩾50, a read depth ⩾20 and a read position rank sum ⩾–8 were selected for the present study. From these, only genetic variants that showed a coverage depth ⩾2 for the alternative allele were included (n = 3,564,615 genetic variants). In order to discard genetic variants that showed a different genotype between the forward and reverse strands, genetic variation showing a Fisher’s strand bias ⩾60 and a strand odds ratio ⩾3 were excluded from the study. In addition, in order to keep only those genetic variants that were confidently mapped, we selected only genetic variation that showed a mapping quality score ⩾40 and a mapping quality rank sum ⩾–10. After QC, only variants showing a minor allele frequency <0.05 were included in downstream analyses.
In order to discard any confounding effect from population stratification in the CD patient cohort, we performed principal component analysis (PCA) incorporating genome-wide association study (GWAS) data from a cohort of 1558 healthy controls from the Spanish population. These individuals were collected and genotyped by the Immune-Mediated Inflammatory Disease (IMID) Consortium, as described previously. 19 After selecting the single nucleotide polymorphisms (SNPs) that were in common between both samples, we evaluated the existence of population stratification using the EIGENSOFT (v4.2) software. 20 Using the first 10 principal components (PCs) of variation over 10 iterations, we did not detect any CD patient showing an outlier genetic background (Figure S1).
Annotation of genetic variation
Low-frequency variants were annotated using the SnpEff software, 21 and their damaging potential was predicted using PolyPhen-2 and Sift.22,23 In PolyPhen-2, the functional significance of allele replacement is predicted by a Naïve Bayes classifier approach that also estimates the probability of each variant to be classified as damaging when it is nondamaging [false positive rate (FPR)]. In the present study, low-frequency variants were defined as damaging if classified as possibly or probably damaging by both PolyPhen-2 (FPR <10% and FPR <20% for probably and possibly damaging, respectively) and Sift, as previously described. 13 Based on the annotation displayed in the Exome Aggregation Consortium (ExAC) Database, 24 damaging variants were characterized according to their impact using the Sequence Ontology tool. 25 Briefly, damaging variants were classified into: start lost: codon variants that change at least one base of the start codon; stop gained: sequence variants that change at least one base of a codon leading to a premature stop codon and shortening of the protein; stop lost: sequence variants that change at least one base of the terminator codon resulting in an elongated transcript; synonymous: sequence variants that do not alter the amino acid sequence encoded; missense: sequence variants that change one or more bases resulting in a different amino acid sequence without altering the length of the protein sequence; splice donor: splice variants that change the two base pair region at the 5′ end of an intron; splice region: sequence variants that change the region of the splice site either within 1–3 bases of the exon or 3–8 bases of the intron; 5′ UTR: variants mapping on the 5′ UTR region; 3′ UTR: variants mapping on the 3′ UTR region; structural interaction: variants that impact the internal interactions of the resulting polypeptide structure; lincRNA: variants mapping on a long intergenic noncoding RNA region; snRNA: variants mapping on a small nuclear RNA region; sense intronic: variants mapping on a noncoding transcript region located within an intronic region with no overlap with exonic sequences; nonstop decay: variants that might impact the translation of mRNA molecules lacking a stop codon; and nonsense-mediated decay: variants that might impact the mRNA degradation.
According to the methodology implemented in SnpEff, genetic variants that correlate with the complete loss of function of the encoded transcript were classified as LoF. These genetic variants include: stop codon-introducing variants, start codon-introducing variants, splice site-disrupting variants, insertion/deletion variants predicted to disrupt the transcript reading frame, and large deletions that remove either the first exon of the gene or more than 50% of the protein-coding sequence of the affected transcript. 21 Given that stop codon-introducing and frameshift variants are enriched toward the 3′ end of the gene and tend to show tolerance to truncation close to the 5′ end, putative LoF variants identified in the last 5% of the coding region were not included in the present study.
For each variant predicted as damaging or LoF, we also estimated its putative impact at the protein level using the method implemented in SnpEff. 26 According to this method, three categories were defined: high impact, the variant is assumed to have a disruptive impact in the protein, probably causing protein truncation, loss of function or triggering nonsense mediated decay; moderate impact, the variant is nondisruptive and might change the protein effectiveness; and low impact, the variant is assumed to be mostly harmless or unlikely to change the protein behavior.
SKAT-O rare variant burden test
The contribution of low-frequency damaging variants from the TNF signaling pathway in the clinical response to anti-TNF therapy was investigated using the SKAT-O set-based collapsing method implemented in EPACTS software. 27 This statistical approach was designed to efficiently unify both the burden and nonburden algorithms for rare variant analysis. While the former method collapses genetic variation into a single burden variable, the latter aggregates the individual variant-score test statistics resulting from the association analysis into a kernel matrix. With this algorithm, the SKAT-O test has been found to maximize the statistical power of the analysis. We used the SKAT-O test to compare the global burden of low-frequency variation at the TNF signaling pathway between responders and nonresponders to anti-TNF therapy. This statistical analysis was adjusted for the clinical variables that showed a significantly different distribution between responder and nonresponder patients (Table 1). The annotation for the TNF signaling pathway was obtained from the Kyoto Encyclopedia of Genes and Genomes (KEGG) reference database (hsa04668, n = 108 genes). 28
Epigenetic enrichment analysis
In order to identify the relevant cell types for anti-TNF response, we performed an epigenetic enrichment analysis. First, we analyzed the enrichment of genome-wide LoF variation (n = 568 SNPs) on cell-type-specific H3K4me3 chromatin marks using the EPIGWAS software. 29 Second, we followed the same procedure to analyze the enrichment of damaging variation at the TNF signaling pathway on cell-type-specific H3K4me3 chromatin marks. Cell type-specific epigenetic data from H3K4me3 was obtained from the Roadmap Epigenomics project. 30
The statistical methodology implemented in the EPIGWAS software consists of five consecutive steps. First, the LD between the associated variants and those located within a distance window of 500 kb is computed to define the loci to be analyzed. Second, the regulatory activity of each variant in each cell type is scored as the height of the nearest H3K4me3 peak, divided by the distance between the SNP and the summit of the peak. For each cell type and locus, the value of the variant with the highest score is selected as a measure of the overlap between the locus and the cell type-specific regulation. Third, these scores are normalized using Euclidean methodology. Fourth, the scores from all loci are summed within each cell type in order to compute the cell type regulatory score. In the last step, the statistical significance of cell-type-specific overlap was obtained using a permutation-based approach (n = 10,000 permutations). In each permutation, a matched set of SNPs is selected from a set of independent SNPs from the HapMap Project and the cell type specific regulatory score is computed again for each cell type. The statistical significance of the enrichment analysis is finally defined as the proportion of sets of SNPs matched with a cell type specific regulatory score exceeding the observed score for the associated variants of interest.
Results
Study population
A total of 41 CD patients were included. The main characteristics of the study cohort are summarized in Table 1. Mean CDAI was 256 (standard deviation = 57). Most of the patients had ileocolonic location and inflammatory behavior, and over 40% had been operated on due to CD. Approximately 40% received adalimumab and 60% infliximab. At week 14, 25 patients (61%) achieved remission and 6 had partial response (14.6%), and therefore considered responders, and 10 (24.4%) were primary nonresponders. When comparing the distribution of the different epidemiological and clinical variables between responders and nonresponders to anti-TNF therapy, we detected significant differences only in the disease behavior (p < 0.05, Table 1). In addition, there was no difference in CDAI score at baseline between responders and nonresponders (259 versus 246, p = 0.5).
Classification of low-frequency damaging variants
A total of 14,994,115 genetic variants were identified by WGS, of which 1,738,956 were found to be low-frequency variants. Using PolyPhen-2 and Sift, 2682 low-frequency genetic variants were predicted to be damaging. In order to characterize the effect of the damaging variants, we classified them according to their damaging potential (Table 2). Overall, we found that 1.9% and 98.1% of the low-frequency damaging variants have a high and moderate/low impact at the protein level, respectively.
Characterization of the damaging low-frequency genetic variation identified in the study population.

Number of low-frequency damaging variants showing the indicated annotation. Given that a single genetic variant can impact different genes, the annotated variants outnumber those predicted as damaging.
Genome-wide LoF variants associated with anti-TNF therapy response
We detected 568 low-frequency LoF variants associated with response to anti-TNF therapy, including 281 and 386 variants in adalimumab- and infliximab-treated patients, respectively (n = 99 overlapping variants). A total of 275 and 384 low-frequency LoF variants were detected in patients with and without prior surgery, respectively (n = 92 overlapping variants). They were all heterozygous LoF except for two variants that had homozygous LoF carriers. These variants mapped to the HLA-B (SNP rs41563412) and HLA-DRB1 (SNP rs1071752) genes. The unique homozygous carrier of the damaging HLA-B rs41563412-GCA variant (frameshift mutation, p.Ser33LeufsTer9) did not respond to anti-TNF therapy (i.e. adalimumab) and was not involved in a surgical intervention. Conversely, the three patients that carried one or two copies of the damaging HLA-DRB1 rs1071752-C variant (synonymous mutation, p.Gln125Gln) were all responders to anti-TNF therapy (i.e. infliximab), and only one of them was not involved in a surgical intervention. Both SNPs were found to be protein-coding variants with a high functional impact at the protein level. The strongest damaging impact was detected in 10 LoF SNPs mapping at NECAP2, HEYL, THNSL2, NPHP3-ACAD11, CLDN18, LY6G6C, C6orf226, RAB2IL1 and EFCAB5 genes (Table 3). The SNP chr2:88173152 at THNSL2 gene was not annotated in the ExAC or dbSNP databases, 31 suggesting this could be a new genetic variant.
Characterization of the damaging loss-of-function variants identified in the study population.

A1, reference allele; A2, alternative allele that causes the LoF of the protein encoded by the indicated gene; CD, number of CD patients carrying the LoF variant; Chr, chromosome; Coord, SNP coordinates in GRCh38/hg38; Nresp, number of patients that carry the LoF variant and did not show a significant clinical response to anti-TNF therapy; Resp, number of patients that carry the LoF variant and responded to anti-TNF therapy; SNP, single nucleotide polymorphism.
Using cell-type epigenetic enrichment analysis, we found that the genome-wide LoF variants associated with anti-TNF response were significantly enriched for epigenetic marks characteristic of the colonic (p = 4.11e–4) and duodenum mucosa (p = 0.011, Figure 1). In patients with prior surgery, the genome-wide LoF variants associated with the overall anti-TNF response were significantly enriched in epigenetic marks from the CD34+ hematopoietic precursor cells (p = 0.021), whereas a significant enrichment in epigenetic marks from the colonic (p = 4.10e–4) and duodenum mucosa (p = 7.61e–3) was detected in patients without prior surgery. When performing the cell-type epigenetic enrichment analysis stratified by drug, the infliximab-associated LoF variants showed also a significant enrichment in epigenetic marks from the colonic mucosa (p = 6.01e–4) as well as from CD34+ hematopoietic precursor cells (p = 0.012). The adalimumab-associated LoF variants showed a significant enrichment in epigenetic marks from the colonic mucosa (p = 2.81e–3), colonic smooth muscle (p = 0.02), CD4+ T cells (p = 0.034) and CD34+ hematopoietic precursor cells (p = 0.043).
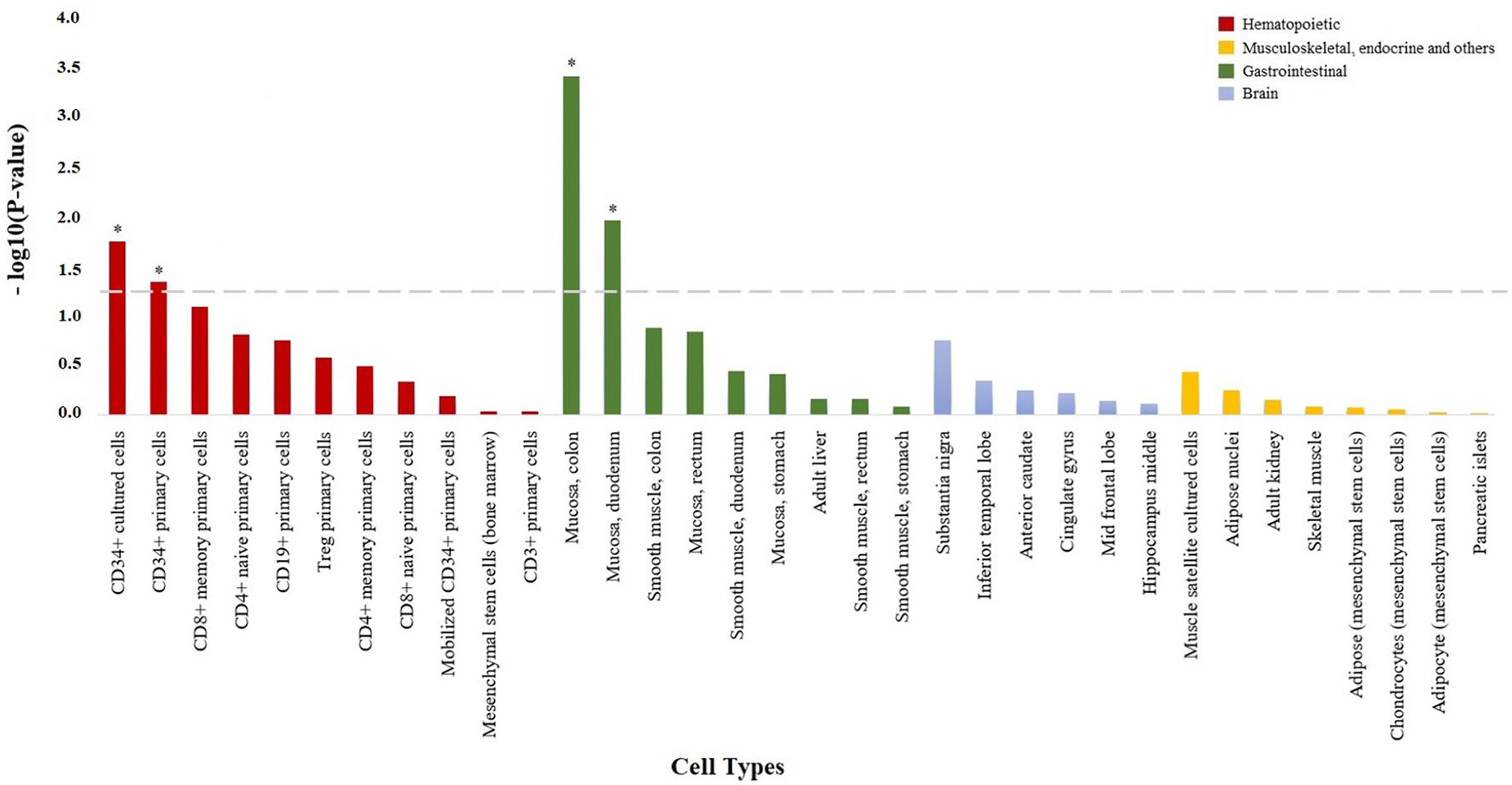
Enrichment of genome-wide loss-of-function variation in H3K4me3 histone marks from specific cell types. Bar plot showing the statistical significance of the overlap between genome-wide loss-of-function variation and H3K4me3 histone marks from the 34 cell types analyzed. All cell types are shown in the x axis, and those with H3K4me3 histone marks significantly enriched in genome-wide loss-of-function variation are represented with an asterisk (p < 0.05).
Association of damaging variation at the TNF signaling pathway with anti-TNF response
A total of 19 damaging low-frequency genetic variants were found to map to the TNF signaling pathway (hsa04668). The analysis of this variation using the SKAT-O test showed that the damaging burden is different between anti-TNF responder (partial response or remission) and nonresponder patients (p = 0.016). The cell-type enrichment analysis showed an increase in epigenetic marks specific from immune cell types (Figure 2), principally CD8+ T cells (p = 6.01e–4) but also CD4+ T cells (p = 0.032). Like in the functional characterization of the LoF variants, epigenetic enrichment analysis also indicated an association with gut tissue (colonic mucosa, p = 0.031).
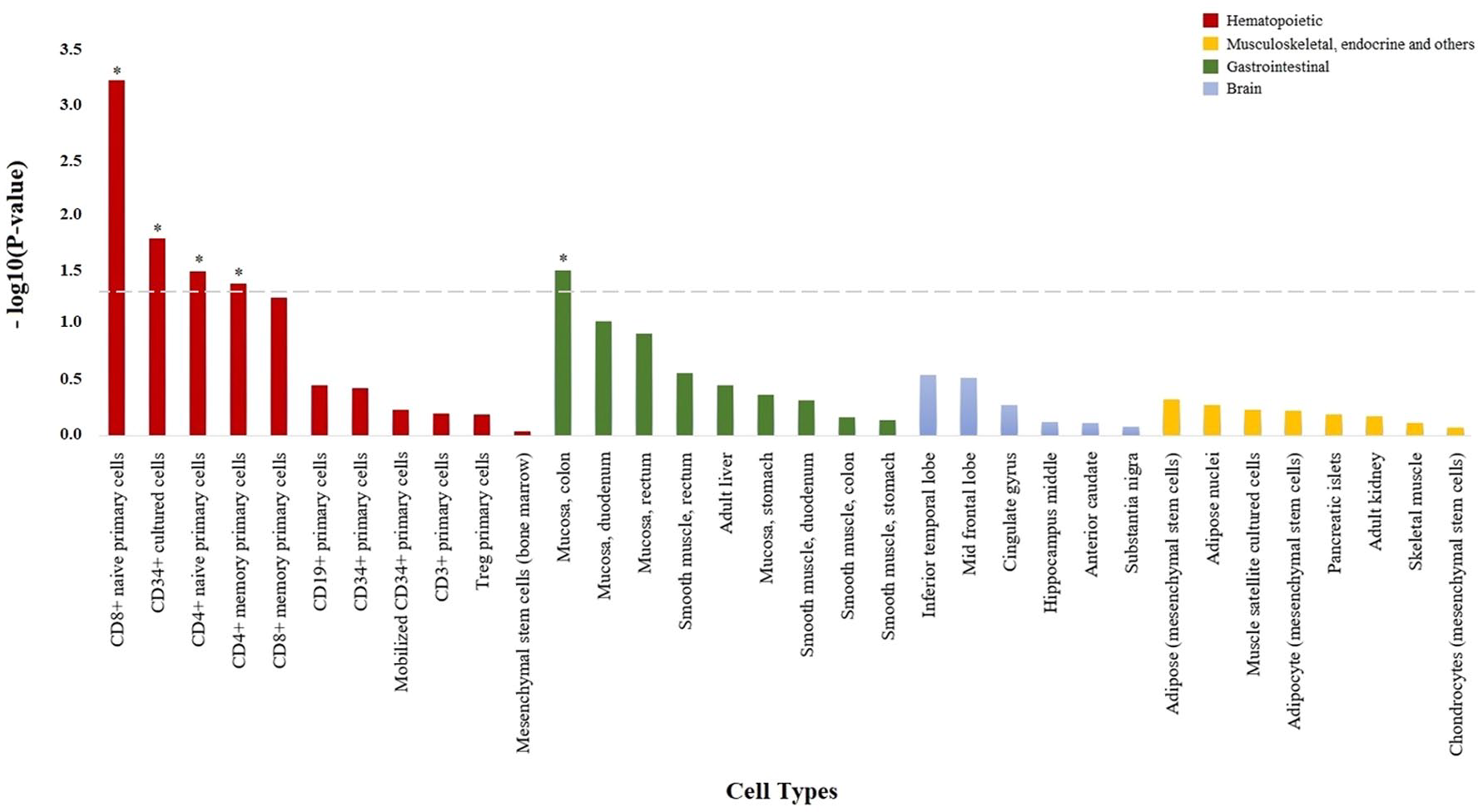
Enrichment of damaging variation at TNF signaling pathway in H3K4me3 histone marks from specific cell types. Bar plot showing the statistical significance of the overlap between damaging variation from the TNF signaling pathway and H3K4me3 histone marks from the 34 cell types analyzed. All cell types are shown in the x axis, and those with H3K4me3 histone marks significantly enriched in damaging variation from the TNF signaling pathway are represented with an asterisk (p < 0.05).
The analysis of low-frequency genetic variants mapping to the TNF signaling pathway using the SKAT-O test showed that the damaging burden is different between anti-TNF responder (partial response or remission) and nonresponder patients (p = 0.016). When stratifying the analysis by anti-TNF agent, we detected that the burden of damaging variation is significantly different between responders and nonresponders to infliximab therapy (p = 9.76e–3). No significant differences were detected among adalimumab-treated patients (p > 0.05). The damaging burden was found to be also significantly different between responders and nonresponders to anti-TNF therapy in patients without prior surgery (p = 0.011), but not among those patients that had not been previously involved in a surgical intervention (p > 0.05).
Discussion
In this study, we sequenced the whole genome of CD patients to examine the hypothesis that functional rare variants are associated with anti-TNF response. Using genome-wide and set-based analyses, we found evidence of LoF and damaging variants influencing the anti-TNF response. Cell type enrichment analysis suggests that these variants affect the cytotoxic response of the adaptive immune system (CD8+ naïve T-cells) and the gastrointestinal mucosa.
A major finding from this study is that, among the millions of variants of the human genome, the only two homozygous LoF SNPs were found to map to HLA genes. These two variants, rs1071752 and rs41563412, are located in an exonic region of the HLA-DRB1 gene and an intronic region of the HLA-B gene, respectively. The proteins encoded by the HLA-DRB1 and HLA-B genes play a central role in the adaptive immune system by presenting antigens to T-cells, including CD4+ [in a major histocompatibility complex class II (MHC)-II dependent manner as is the case of HLA-DRB1] and CD8+ T-cells (in an MHC-I dependent manner as the HLA-B), being the adaptive immune response altered in CD patients.32–34 Although it has been shown that genetic factors underlying clinical phenotypes of CD (i.e. other than anti-TNF response) might not necessarily be shared with CD risk loci, the HLA region itself is indeed one of the strongest risk loci for CD, and multiple genetic association studies have previously identified variation at HLA-DRB1 and HLA-B genes showing a significant association with CD risk.35–38 To our knowledge, genetic variation at these two genes has not been previously associated with anti-TNF response in CD. Our results support that genetic variation at the HLA-B and HLA-DRB1 disease susceptibility loci could also modulate anti-TNF efficacy, and, therefore, these two genes could be two promising biomarkers for future tailored TNF-blocking therapy in CD patients.
In the genome-wide screening for LoF variants, we have also identified functionally-relevant variants at nine genes, including NECAP2, HEYL, THNSL2, NPHP3-ACAD11, CLDN18, LY6G6C, C6orf226, RAB3IL1, and EFCAB5. CLDN18 encodes a tight junction protein from the claudin family that regulates the integrity of the intestinal epithelium. 39 The transcriptomic profile of claudins has been found to depend on disease activity, and to vary along the intestinal mucosa. 40 CLDN18 has been recently associated with the pathogenesis of IBD, 35 where, by modulating the permeability of the epithelial barrier, it may allow the lamina propria immune system to encounter luminal antigens that otherwise would not be found. Of relevance, the LY6G6C gene maps to a cluster of leukocyte antigen genes located in the MHC-III. 41 The MHC-III locus also contains the TNF gene, indicating that this genomic region is likely to be important in regulating the inflammatory activity of immune-mediated diseases. 42 Given the physical proximity between the LY6G6C and TNF genes (i.e. ~140 kb), our results suggest that variation at LY6G6C could alter the expression of the TNF gene, and, consequently, influence the anti-TNF response.
We also found that the LoF variant at THNSL2 gene (i.e. chr2:88173152 SNP) is not annotated in the databases of reference for genetic variation. 31 The THNSL2 gene encodes a threonine synthase-like protein. To date, however, its biological function in humans is still unknown. The scarce information on the biology of the THNSL2 gene derives from an in vitro study showing that THNSL2 is transcribed into a rare mRNA splice-variant that leads to SOAFT formation, a human T cell secreted cytokine that exacerbates the inflammatory state of autoimmune diseases. 43 In our study cohort, the nonresponse phenotype of the patient carrying the chr2:88173152 SNP suggests that this variant could have a functional effect on the anti-TNF mechanism of action that compromises the treatment response. Future targeted sequencing studies on the THNSL2 gene are warranted to confirm the involvement of the chr2:88173152 SNP TNF signaling in CD.
Epigenetic mark enrichment analysis of the LoF variants indicates that the gut mucosa is an important mediator of anti-TNF response in CD. Previous genetic studies have shown the importance of the colonic mucosa in the regulation of the inflammatory processes that characterize the pathogenesis of CD. 4 The gastrointestinal mucosa of CD patients is highly enriched in activated TNF-secreting macrophages.44–46 From a pharmacological perspective, the anti-TNF therapy has been shown to inhibit not only the gut inflammation but also the inflammation-induced angiogenesis in the intestinal mucosa.47,48 Therefore, our findings are consistent with the critical role of the intestinal mucosa as the location where the immune response is initiated in CD, and, therefore, as the key organ determining the clinical response to anti-TNF therapy.
In this study, we provide the first evidence of association between rare damaging variation in the TNF signaling pathway and anti-TNF response. A recent sequencing study in rheumatoid arthritis found that rare variants in this pathway do not contribute to treatment response. 13 However, in this latter study, only exomes were sequenced, and, thereby, important functional variation from other genomic regions was missed. Additionally, there is clear evidence that rheumatoid arthritis and CD have very little genetic risk overlap. This also suggests that different biological mechanisms operate through TNF signaling, and, consequently, drug efficacy is mediated by different groups of genes and associated regulatory variants.
The epigenetic enrichment analysis of the TNF signaling pathway indicated that CD8+ T-cells are important determinants of the efficacy of anti-TNF agents. The inflamed mucosa of CD patients carries large numbers of immune cells, including CD8+ T cells, 49 which are the main cell type responsible for destroying the intestinal epithelium, leading to an enhanced exposure to the gut microbiota and the subsequent triggering of chronic inflammatory processes.50–52 As opposed to CD4+ T-cells, antigen presentation to CD8+ T-cells is MHC-I restricted. Indeed, the rs41563412 variant located in an intronic region of the HLA-B gene is included within such an MHC-I family. Besides, the unique homozygous carrier of the damaging HLA-B rs41563412-GCA variant (frameshift mutation, p.Ser33LeufsTer9) did not respond to anti-TNF therapy, suggesting a role of this LoF SNP in modulating the CD8+ cytotoxic immune response. The relation between CD8+ T-cells and the response to anti-TNF therapy has been also investigated in previous experimental studies. 46 Using in vitro isolated CD8+ T cells, the administration of anti-TNF therapy was shown to inhibit the proliferation of this T cell subtype both in the gut mucosa and in blood.53,54 Importantly, the transcriptomic profile of CD8+ T cells has been associated with the incidence of being refractory to disease treatment. 55 Notably, in our study, the relationship between CD8+ T-cells and anti-TNF response was restricted to the naïve, but not the memory, subset of CD8+ T-cells, suggesting a primary defect on this cell fraction. Our findings are in line with these results, and support the predominant role of CD8+ T cells in the TNF-dependent inflammatory activity in the gut.
Our study has some limitations. First, the response was categorized based on clinical activity indexes. However, we included patients with high activity index scores to ensure that the symptoms were due mainly to the activity of the disease. In fact, in clinical practice, short-term response is commonly based on patient symptoms. The sample size of the present study is low if compared with genome-wide studies on common variants. However, compared with GWAS, here, we used WGS to identify all possible rare variants. Subsequently, by focusing our analysis only on rare variants with great functional impact, we increase the power of our study. In this respect, this strategy is similar to that used to identify variations associated with rare Mendelian diseases, where a small number of patients have almost exclusive mutations. In this regard, our findings are not trivial at all, as among all genes, the only homozygous LoF homozygosity, a topmost damaging variation, occurred by chance only in HLA genes, which encode key antigen-presenting proteins and are known risk factors in IBD. Finally, given our limited sample size, we restricted rare variation burden analysis specifically to the anti-TNF pathway, thereby preventing a large penalty by testing and analyzing hundreds of other biological pathways.
The analytical strategy implemented in this study is a distinct feature of the present work. In order to identify rare damaging variants underlying the anti-TNF response, we screened the whole genome of anti-TNF-treated CD patients. Compared with array-based genome-wide studies, WGS efficiently captures rare variants that otherwise would have been missed. 56 The analytical approach used for testing association of the TNF signaling pathway is also radically different from single-marker tests. Single-marker tests are underpowered to detect significant associations of low-frequency variants, unless very large sample sizes are used. 57 The set-based collapsing analysis method used here, SKAT-O, groups low-frequency variants into gene sets in order to detect significant overabundance of low-frequency variation associated with complex traits at high statistical power, despite using relatively small cohorts. 27 Given the sample size of our study, we restricted the analysis to the key signaling pathway that is interfered with by anti-TNF drugs. Using this approach, we found a significant burden of rare damaging variants associated with the response to TNF alpha blockers in CD.
In conclusion, we report evidence that functional low-frequency variation is associated with the response to anti-TNF therapy in CD. These findings also provide new insights into the underlying heterogeneity of CD, revealing the basis of TNF-dependent biological mechanisms. The identification of this diversity at the molecular level will be essential to allow the stratification of patients, and, ultimately, help clinicians choose the most appropriate drug.
Supplemental Material
Figure_S1 – Supplemental material for Functional rare variants influence the clinical response to anti-TNF therapy in Crohn’s disease
Supplemental material, Figure_S1 for Functional rare variants influence the clinical response to anti-TNF therapy in Crohn’s disease by María Chaparro, Adrià Aterido, Iván Guerra, Marisa Iborra, Jose Luis Cabriada, Luis Bujanda, Carlos Taxonera, Valle García-Sánchez, Ignacio Marín-Jiménez, Manuel Barreiro-de Acosta, Isabel Vera, Maria Dolores Martín-Arranz, Borja Hernández-Breijo, Francisco Mesonero, Laura Sempere, Fernando Gomollón, Joaquín Hinojosa, Fernando Bermejo, Belén Beltrán, Ainhoa Rodríguez-Pescador, Jesús María Banales, David Olivares, Patricia Aguilar-Melero, Luis Menchén, Rocío Ferreiro-Iglesias, Isabel Blazquez Gómez, Beatriz Benitez García, Luis G Guijarro, Alicia C Marin, David Bernardo, Sara Marsal, Antonio Julia and Javier P Gisbert in Therapeutic Advances in Gastroenterology
Footnotes
Acknowledgements
María Chaparro and Adria Aterido contributed equally as first authors. Antonio Julia and Javier Gisbert contributed equally as senior authors. The authors thank Dr Simon Heath and Dr Raul Tonda from the Centre Nacional d’Anàlisi Genòmica (CNAG, Barcelona, Spain) for their help in the WGS data processing.
Author contributions
María Chaparro and Javier P. Gisbert: study design, data collection, data analysis, data interpretation, writing the manuscript.
Antonio Julià and Adrià Aterido: Genetic analysis, data analysis, data interpretation and manuscript writing.
Borja Hernández-Breijo, Luis González Guijarro, Alicia C. Marín and David Bernardo: Sample management.
Remaining authors: Study design, patient inclusion, sample obtaining and data collection.
All authors approved the final version of the manuscript.
María Chaparro and Javier P. Gisbert are the guarantors of the article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by grants from the “Instituto de Salud Carlos III” (FIS.12/02557 and PI13/00041).
Conflict of interest statement
M Chaparro has served as a speaker, or has received research or education funding from MSD, Abbvie, Hospira, Pfizer, Takeda, Janssen, Ferring, Shire Pharmaceuticals, Dr Falk Pharma, Tillotts Pharma.
I Guerra has served as a speaker for Pfizer and Janssen and consultant and advisory member for Kern Pharma and MSD.
M Iborra has received funding for educational activities, research projects and scientific meetings from Takeda and Abbvie.
JL Cabriada has served as consultant for and received research funding from MSD, Abbvie, Pfizer and Kern Ph.
L Bujanda: none
C Taxonera has served as a speaker, a consultant and advisory member for, or has received research funding from, MSD, Abbvie, Hospira, Pfizer, Takeda, Ferring, Faes Farma, Shire Pharmaceuticals, Dr Falk Pharma, Gebro Pharma.
V García-Sánchez has served as a speaker, a consultant and advisory member for or has received research funding from MSD, Abbvie, Hospira, Kern Pharma, Takeda, Pfizer, Ferring, Faes Farma, Shire Pharmaceuticals, Dr Falk Pharma, Janssen, Gebro Pharma and Otsuka Pharmaceutical.
I Marín-Jiménez has served as a consultant, advisory member, speaker, or has received research funding from Abbvie, Chiesi, Faes Farma, Falk-Pharma, Ferring, Gebro Pharma, Hospira, Janssen, MSD, Otsuka Pharmaceutical, Pfizer, Shire, Takeda, Tillots, and UCB Pharma.
M Barreiro-de Acosta has served as a speaker, a consultant and advisory member for or has received research funding from MSD, Abbvie, Hospira, Pfizer, Kern Pharma, Biogen Takeda, Ferring, Faes Farma, Shire Pharmaceuticals, Dr Falk Pharma, Tillotts Pharma, Chiesi, Gebro Pharma, Otsuka Pharmaceutical and Vifor Pharma.
I Vera has served as a speaker for AbbVie, Takeda and Shire
MD Martín-Arranz has served as a speaker, a consultant and advisory member for MSD; Abbvie, Takeda, Janssen, Ferring, Faes Farma, Shire Pharmaceutical, Tillots Pharma and Chiesi
B.Hernández-Breijo: None
F.Mesonero: None
L Sempere has served as a speaker, a consultant and advisory member for TAKEDA, ABBVIE, KERN, MSD Y TILLOTTS.
F Gomollón has received fees for lectures from Janssen, Abbvie, Takeda, MSD and research grants (Group): MSD, Abbvie. Advisory Committees: None active at present.
J Hinojosa Abbvie, MSD, Takeda, Janssen, Kern, Pfizer-Hospira, Otsuka, Faes, Ferring, Biogen, Shire.
F Bermejo has served as a speaker, a consultant and advisory member for, or has received research funding from, MSD, Abbvie, Pfizer, Hospira, Takeda, Ferring, Faes Farma, Shire, Tillotts, Chiesi and Gebro.
B Beltrán: None
A Rodríguez Pescador
JM Banales: has served as a speaker and advisory member for OWL Metabolomics.
D Olivares: None
P Aguilar-Melero: None
L Menchén has served as a speaker, a consultant and advisory member for or has received research funding from MSD, Abbvie, Hospira, Pfizer, Takeda, Ferring, Faes Farma, Shire Pharmaceuticals, Dr Falk Pharma, Tillotts and General Electric.
R Ferreiro-Iglesias has received grants from Abbvie,MSD, Shire, Falk, Tillotts.
I Blazquez Gomez: None
B Benítez García: None
LG Guijarro: None
AC Marin: None
D Bernardo: None
JP Gisbert has served as a speaker, a consultant and advisory member for, or has received research funding from, MSD, Abbvie, Hospira, Pfizer, Kern Pharma, Biogen, Takeda, Janssen, Roche, Ferring, Faes Farma, Shire Pharmaceuticals, Dr Falk Pharma, Tillotts Pharma, Chiesi, Casen Fleet, Gebro Pharma, Otsuka Pharmaceutical, Vifor Pharma.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.