
Abstract
Nonalcoholic fatty liver disease (NAFLD) represents a spectrum of hepatic pathology, ranging from simple accumulation of fat in its most benign form, steatohepatitis, to cirrhosis in its most advanced form. The prevalence of NAFLD is 20–30% in adults, and 10–20% of patients with NAFLD progress to nonalcoholic steatohepatitis (NASH) which is predicted to be the leading cause of liver transplantation over the next 10 years. Therefore, it is essential to explore effective diagnostic and treatment strategies for NAFLD patients. Chemokines are a family of small and highly conserved proteins (molecular weight ranging from 8 to 12 kDa) involved in regulating the migration and activities of hepatocytes, Kupffer cells (KCs), hepatic stellate cells (HSCs), endothelial cells and circulating immune cells. Accumulating data show that chemokine and its receptor act vital roles in the pathogenesis of NAFLD. Herein, we summarize the involvement of the chemokine and its receptor in the pathogenesis of NAFLD and explore the novel pharmacotherapeutic avenues for patients with NAFLD.
Introduction
Nonalcoholic fatty liver disease
Nonalcoholic fatty liver disease (NAFLD), firstly described in 1980 by Ludwig et al., 1 is becoming the most frequent chronic liver disease worldwide, and morbidity of NAFLD is up to 20–30% in adults and even higher among obesities and type II diabetes mellitus patients.2–7 NAFLD represents a spectrum of hepatic pathology, ranging from simple accumulation of fat (‘fatty liver’ or steatosis) in its most benign form, to steatohepatitis, to cirrhosis in its most advanced form.8,9 NAFLD is regarded as the hepatic manifestation of the metabolic syndrome, including central obesity, insulin resistance, dyslipidemia, and hypertension. Nonalcoholic steatohepatitis (NASH) is mediated by inflammatory cytokines, mitochondrial dysfunction secondary to nutrient excess and oxidative stress,10,11 resulting in hepatocyte infla-mmation, ballooning, apoptosis, and activation of hepatic stellate cells (HSCs). About 10–20% of patients with NAFLD develop NASH, 12 which is predicted to become the leading cause of liver transplantation over the next decades. 13
Pathogenesis of NAFLD
Nearly 20 years ago, the ‘two hit’ hypothesis was proposed. The ‘first hit’ refers to hepatic steatosis which is a benign form, while the ‘second hit’ consists of oxidative stress and inflammation that leads to severe liver damage. 14 In recent years, the ‘multiple hit’ hypothesis has been put forward for interpreting the progression of NAFLD. As shown in Figure 1, in addition to metabolic factors, innate immune alterations, inflammation response caused by free fatty acids (FFAs), bacterial lipopolysaccharide (LPS), chemokines, cytokines, and adipokines are implicated in the development of NAFLD-NASH hepatocellular carcinoma (HCC).12,15–20 Other organs such as adipose tissue, muscle, and intestine also participate in the pathogenesis of NAFLD, and aggravate its systematic metabolic disorder. 10

The ‘multiple hit’ hypothesis for the progression of NAFLD and natural history of NAFLD: dietary habits, environmental and genetic factors result in the development of insulin resistance, obesity with adipocyte proliferation and changes in the gut microbiome. Insulin resistance leads to increased hepatic DNL and dysfunction of adipose tissue and adipocyte. Fat accumulates in the liver in the form of TG. Contemporarily, this happens with increased lipotoxicity from high levels of FFAs, free CH and other lipid metabolites, which induce mitochondrial dysfunction with oxidative stress and production of ROS and ER stress with activation of UPR. All these factors lead to hepatic inflammation, apoptosis and fibrosis. Also, the altered gut microbiome results in more production of FFAs and CH and increases small bowel permeability. Increased fatty acid absorption causes the activation of inflammasome such as LPS, and the release of proinflammatory cytokines such as IL-6 and TNF-α.
Chemokines and NAFLD
Chemokines, a family of small and highly conserved proteins (with molecular weights ranging from 8 to 12 kDa), are involved in multiple biological processes, including chemotaxis, 21 leukocyte degranulation, 22 hematopoiesis 23 and angiogenesis, 24 and mediate immune cell trafficking and lymphoid tissue maturation.25,26 Although the chemokines have a highly conserved and three-stranded β-sheet/α-helix tertiary structural fold, their quaternary structures vary as compared with their subfamily. As indicated in Figure 2, according to the sequential positions of the first two cysteine residues, chemokines are divided into four main subtypes: CC-chemokines, CXC-chemokines, C-chemokines and CX3C-chemokines,26,27 and possess tiny differences in structure. In CC and CXC chemokines, the first two cysteines are adjacent (CC motif) or separated by one amino acid residue (CXC motif). C chemokines lack the first and third of the cysteines, and CX3C chemokines have three amino acids between the first two cysteine residues.27,28 In terms of the chemokine specific binding with different receptors and vice versa, a superfamily of chemokines has been presented26,28–30 (Table 1). As shown in Figure 3, the chemokine receptors, 7-transmembrane G protein-coupled receptors consist of an N-terminus outside the cell surface, three extracellular and three intracellular loops as well as a C-terminus in the cytoplasm.28,31 The interaction between chemokines and their receptors has a key role in the occurrence of liver diseases.

The structure of chemokines. The structures of chemokines are similar with at least three β-pleated sheets (indicated as β1-3) and a C-terminal a-helix. In the CXC chemokine family, the two cysteines nearest the N-termini are separated by a single and variable amino acid. In the CC chemokine family, the two cysteines nearest the N-termini are adjacent. In the C chemokine family, only one cysteine is near its N-terminus. The CX3C chemokine has a typical chemokine-like structure. The two cysteines nearest the N-termini are separated by three amino acids. In addition, the chemokine domain occurs at the end of a long stalk which is largely replaced by mucin-like carbohydrates. The protein is fixed in the membrane and has a short cytoplasmic domain.
The classification of chemokines and their receptors.


The structure of the typical chemokine receptor. Chemokine receptors are embedded in the lipid bilayer of the cell surface and possess seven transmembrane domains.
The liver tissues are enriched by various kinds of immune cells including Kupffer cells (KCs), resident macrophages, natural killer (NK) cells and other innate immune cells, such as neutrophils, leukocytes, monocytes, and inflammatory macrophages. 32 The adaptive immune system is primarily composed of natural killer T (NKT) cells, B-cells, and T-cells. Thus, immune and inflammatory cells have a central role in the pathogenesis of NAFLD. Steatosis, as the critical stage in the progression of NAFLD, is mediated by hepatic inflammation, 8 and chemokines are key players in regulating the migration and activities of hepatocytes, HSCs, KCs, endothelial cells, and circulating immune cells. Chemokines and their receptors recruit immune and nonimmune cells into the inflamed sites. In the liver, resident hepatic macrophages and KCs are important sensors of tissue injury, and depletion of KCs prevents the development of hepatic steatosis and insulin resistance of NASH.33,34 In addition, resident hepatic macrophages and KCs are predominantly activated by pathogen-associated molecular patterns from invading pathogens, and danger-associated molecular patterns released from injured hepatocytes, cholangiocytes, and stellate cells in response to danger signals. Once activated, KCs release proinflammatory cytokines and chemokines, initiate the acute phase response and attract the neutrophils that produce cytotoxic and antimicrobial molecules [reactive oxygen species (ROS), oxidants, defensins]. NKT cells in turn perpetuate inflammatory responses via inflammatory monocytes which differentiate into proinflammatory hepatic macrophages. 35 Therefore, various chemokines, their receptors and chemokine signaling pathways take part in the pathogenesis of NAFLD (Table 2).
Chemokines and their receptors in NAFLD.

HCC, hepatocellular carcinoma; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis.
CC chemokine and its receptor
CCL2 (also termed MCP-1) and its receptor CCR2 are characterized in many chemokines involved in hepatic diseases including NASH, fibrosis and HCC. CCL2, mainly produced by KCs and HSCs, is conductive to inflammation, fibrosis, and steatosis, 35 and recruits macrophages and monocytes into the liver, resulting in the activation of HSCs.37–39 CCR2 is also expressed in HSCs and macrophages. During the progression of NASH, activation of CCL2–CCR2 axis promotes macrophage accumulation, inflammation responses, fibrosis, and steatosis in the liver as well as in adipose tissue, 40 and contributes to NASH by recruiting bone marrow-derived monocytes. 39 In contrast, CCR2-deficient mice display a decreased accumulation of inflammatory cells in the liver,39,41 and the inhibition of the CCL2–CCR2 axis causes reduced hepatic fibrosis in experimental fibrosis models.37,39 Prop-agermanium, as a CCR2 inhibitor, is reported to prevent insulin resistance, steatosis42,43 and NASH. 44 The CCL2–CCR2 axis also mediates the liver injury to lead to parenchymal cell necrosis or apoptosis by release of ROS and activation of HSCs and macrophages. 45 It has been reported that patients with HCC have a lower serum level of CCL2 than cirrhotic patients. 46
Other CC-chemokines and their receptors also participate in the progression of NAFLD. The expression of CCL5 and its receptors CCR1 and CCR5 is increased in liver macrophages and HSCs and facilitates liver fibrogenesis. 49 CCL5 secreted by HSCs directly induces steatosis and proinflammatory factors in healthy hepatocytes through interacting with its receptor, CCR5. 50 CCR1 and CCR5 mediate profibrogenic effects in bone marrow-derived cells and resident liver cells, and promote the migration of HSCs through a redox-sensitive and PI3K-dependent pathway. 51 CCL3 (MIP-1α) and CCL4 (MIP-1β) are upregulated in mouse liver fibrogenesis models and human fibrosis samples, favor mouse liver fibrosis via increasing proliferation and migration of HSCs, and associate with stellate cell activation and liver immune cell infiltration. 48 Inhibition of CCR1 and (or) CCR5 represses the progression of liver fibrosis in mice.51,52 Moreover, pharmacologic inhibition of CCL5 facilitates fibrosis regression in mice. 52 Beyond that, the serum levels of CCL4 and CCL5 are higher in HCC patients than those in cirrhotic patients, indicating that CCL4 and CCL5 as inflammatory chemokines are used to predict the presence of HCC. 46 Additionally, CCL1-CCR8 and CCL25-CCR9 pathways also play critical roles in promoting liver fibrosis by recruiting macrophages.36,59
Recent studies have shown that the expression of CCL20 is increased in patients with NAFLD fibrosis. 53 CCL20 and its exclusive receptor CCR6 contribute to cholestatis and HCC. Accumulation of immune cells in adipose tissue (AT) causes obesity-associated inflammation, insulin resistance and fatty liver. Initially, macrophage-mediated invasion of AT is related to a release of soluble mediators by fat-laden adipocytes 91 and the upregulation of chemokine expression. 92 It is found that mature adipocytes release CCL20 and stimulate the migration of AT lymphocytes via CCR6. 54 CCL20 exerts the tumor-promoting effects by inducing cell migration and epithelial-mesenchymal transition (EMT) via PI3K/AKT and Wnt/β-catenin pathways and predicts poor survival and tumor recurrence in patients with HCC. 55 Moreover, HIF-1α-CCL20-indoleamine 2, 3-dioxygenase (IDO) axis accelerates tumor metastasis by inducing EMT and an immunosuppressive tumor microenvironment. 56 CCL20 also enhances the growth of HCC cells via phosphorylation of p44/42 MAPK. 57 CCR6 possesses elevated expression on the HuH7 cells, resulting in spontaneous secretion of CCL20, 57 and CCL20–CCR6 axis mediates the migration of circulating FoxP3(+) Tregs into tumor microenvironment. 58
CXC chemokine and its receptor
Several CXC-chemokines and their receptors are involved in the progression of NAFLD. Patients with HCC have a higher serum level of CXCL5 than cirrhotic patients. 46 Increased expression of CXCL5 is correlated with neutrophil infiltration and poor prognosis in patients with HCC. 60 CXCR2, a CXCL5-specific receptor, is at a high level in many cancer cells and is related to tumorigenesis, tumor angiogenesis and metastasis.61–63 The CXCL5–CXCR2 axis gives rise to cell spreading by inducing EMT via activation of the PI3K/Akt/GSK-3β/Snail signaling pathway. 64
CXCL8, known as interleukin (IL)-8, is a proinflammatory CXC chemokine with the properties of tumorigenicity and angiogenesis in cancer. 93 The CXCL8 level is increased in HCC tissues and cell lines, and coexpression of HIF-1α and CXCL8 induces HCC progression and metastasis via activation of the AKT/mTOR/STAT3 pathway, indicating that CXCL8 is a prognostic marker and a potential target for HCC. 65 Macrophages are a major component of tumor stroma involved in HCC progression. Recently, macrophages activated by co-cultured liver cancer cells produce higher levels of CXCL8, induce CXCL8/miR-17 cluster, and promote HCC cell proliferation and metastasis. 66 Additionally, tissue-derived fibroblasts, CCL2, CXCL1, and CXCL8 are upregulated in HCC and accelerate its progression. 47
CXCL10 belongs to the ELR-negative CXC family. In the liver, CXCL10 is mainly secreted by the liver sinusoidal endothelium and hepatocytes in areas of lobular inflammation. The CXCL10 serum level is associated with the severity of lobular inflammation and acts as an independent risk factor for NASH patients. CXCL10 induces inflammation, oxidative stress and fibrosis in experimental steatohepatitis via activation of the nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-κB) pathway. 73 In addition, CXCL10 promotes steatosis by stimulating lipogenesis 74 and cholesterol (FFC) diet-induced macrophage-associated hepatic injury and fibrosis. 75 As a proinflammatory factor, CXCL10 is implicated in each phase of NASH development. 73
CXCL9 serum level is also associated with genotypes and severity of liver fibrosis in patients.67,68 CXCR3 is the receptor of multiple ligands such as CXCL4, CXCL9, CXCL10 and CXCL11, 69 and displays a role in the development of steatohepatitis.70,75 CXCR3 contributes to the expansion of liver inflammation to the parenchyma by inducing endothelial transmigration and T-cell chemotaxis to the liver lobule. 71 Increased expression of CXCR3 is confirmed in human NAFLD liver tissues and experimental steatohepatitis models, while the blockade of CXCR3 reverses the established steatohepatitis. 70 CXCR3 promotes cholesterol-induced steatohepatitis and NASH, 75 facilitates lipid accumulation, inflammatory cell infiltration and ER stress, 70 and induces macrophage infiltration and Th1 and Th17 immune response in steatohepatitis.70,72
CXCL11 serum level is increased in patients with liver fibrosis, nonalcoholic cirrhosis and high portal pressure, and is used to predict long-time survival of cirrhotic patients bearing transjugular intrahepatic portosystemic shunts. 76
CXCL12, known as stromal-derived factor 1 alpha (SDF1α) is the ligand of CXCR4 and CXCR7. The CXCL12–CXCR4 axis promotes proliferation, survival, invasion, and metastasis, 77 and mediates interstitial fluid flow to enhance the invasion in HCC cells via the MEK/ERK pathway. 78 CXCL12 activates CXCR4-expressing HCC cells, lymphocytes and endothelial cells in an autocrine or paracrine manner 77 and induces tumor vasculature remodeling under hypoxic conditions. 79 CXCR4 has been demonstrated as upregulated in HCC tissue, 80 but downregulated in MMP10-deficient HCC, while pharmacological inhibition of CXCR4 significantly reduces MMP10-stimulated HCC cell migration. 81 In addition, CXCR7 expression is upregulated in metastatic HCC, and promotes the growth and invasiveness via activation of the MAPK and angiogenesis signaling pathways. 82
CXCL16 is considered to be a survival and maturation factor for patrolling hepatic NKT cells. 83 Hepatic macrophages express a high level of CXCL16 at the early stage of liver injury, thus promoting the rapid accumulation of NKT cells to the injured parts of the intrahepatic tissue. Furthermore, CXCL16-mediated accumulation of NKT cells exhibits the potential to amplify inflammatory signals in the early stage of inflammation, 84 and CXCL16–CXCR6 axis acts in the migration of hepatic NKT cells to promote CCl4-induced liver fibrosis.45,85
CX3C chemokine and its receptor
CX3CL1, the only member of the CX3C subfamily of chemokines is widely expressed in immune cells and nonimmune cells. CX3CR1, a member of 7-transmembrane G protein-coupled receptor is the receptor of CX3CL1. CX3CR1 can be expressed by monocytes, macrophages, microglia, T helper (Th) 1 cells, CD8+T effector/memory cells, NK cells, γδ T-cells, and dendritic cells (DCs). 40 Both CX3CL1 and CX3CR1 are expressed throughout the body, but depend highly on cell type-specific organs and tissues. The CX3CL1–CX3CR1 axis promotes liver fibrosis by activation of HSCs, KCs, biliary epithelial cells and liver infiltrating lymphocytes,86–88 and the interaction between CX3CL1 and CX3CR1 on liver macrophages regulates liver inflammation by enhancing macrophage survival and reducing the anti-inflammatory phenotype.89,90
Application prospect
In terms of the role of chemokines and their receptors in the pathogenesis of liver diseases, some therapeutic strategies for targeting chemokines and their receptors may highlight a new direction for the treatment of patients with liver diseases. As showed in Table 3, pharmaceutical inhibition of CCR2 contributes to an inhibition of liver inflammation and fibrosis by preventing infiltration of Ly6C-positive macrophages. 94 Congruously, previous studies showed that pharmacological inhibition of CCL2 or CCR2 significantly ameliorates insulin resistance, steatosis and inflammation in several metabolic disease models. 95 Of note, cenicriviroc (CVC), a CCR2 and CCR5 antagonist, has the potential to exert anti-inflammatory and antifibrotic activity in animal models. In a phase II study, in obese adults with NASH, CVC does not achieve the ideal effect in the treatment of NAFLD, but shows a reduction in fibrosis scores; a similar result is verified in a phase IIb study in NASH in recent studies.96–98 Moreover, glutaminyl cyclase inhibitors may alleviate liver inflammation by destabilizing CCL2 in an experimental model of NAFLD. 99 Besides that, as a CCR5 antagonist, maraviroc shows the ability in ameliorating the development of hepatic steatosis in a mouse model of NAFLD. 100 Pharmacological inhibition of mixed lineage kinase 3 (MLK3) mediates the release of CXCL10-laden extracellular vesicles from lipotoxic hepatocytes. 101 Anti-CXCL16 decreases liver macrophage infiltration and fatty liver degeneration in NAFLD. 102 These suggest that chemokines and their receptors may act as potential therapeutic targets of NAFLD. Further investigations of the applications of chemokines and their receptors in NAFLD is needed.
Chemokines and their receptors as therapeutic targets for NAFLD treatment.
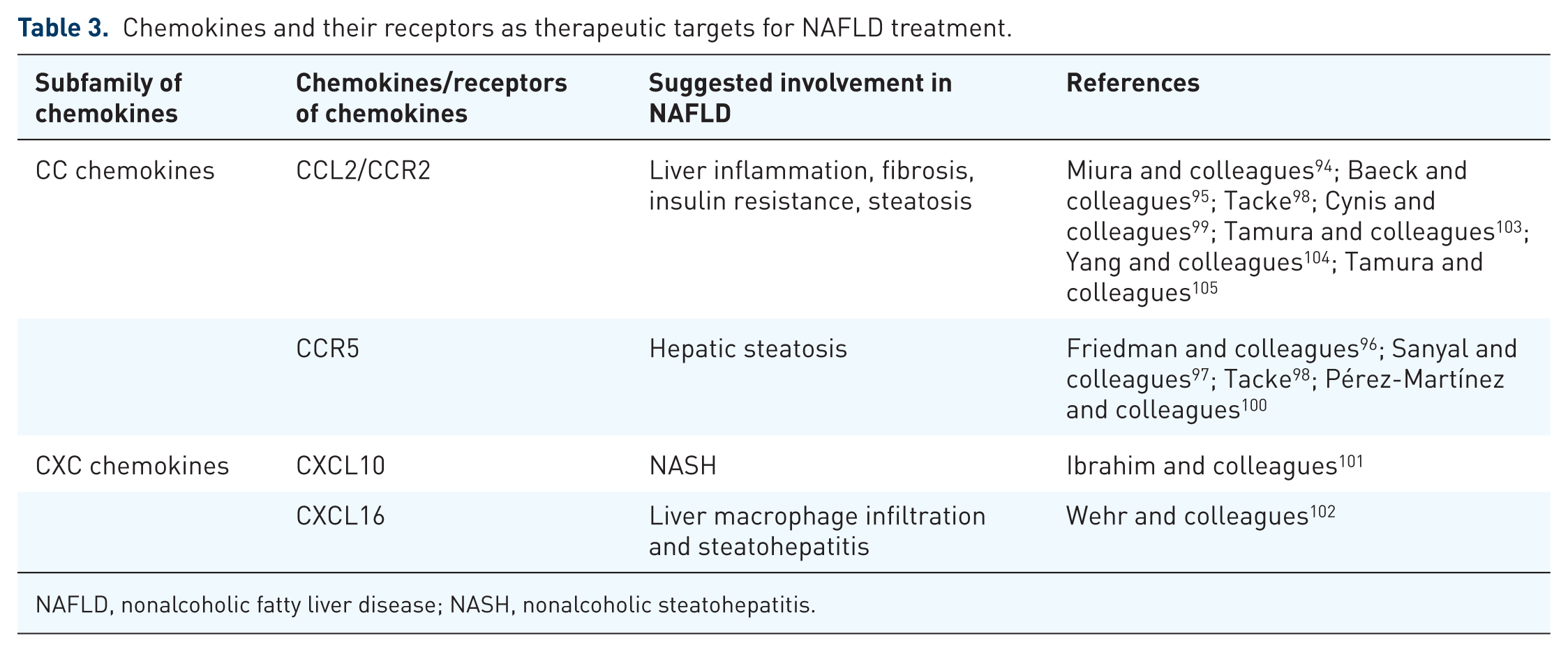
NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis.
Footnotes
Acknowledgements
Wei Chen and Jing Zhang contributed equally to this work.
Funding
This study was supported by grants from the National Nature Science Foundation of China (nos. 81573747 & 81873143), Shanghai Science and Technology Commission Western Medicine Guide project (no. 17411966500), Shanghai Jiaotong University School of Medicine Transformation medicine and Innovation Center research project (no. TM201723) and Shanghai Jiao Tong University School of Medicine doctoral innovation fund (no. BXJ201737).
Conflict of interest statement
The authors declare that there is no conflict of interest.