
Abstract
Background:
Ornithine phenylacetate (OP) has been proven effective in lowering ammonia plasma levels in animals, and to be well tolerated in cirrhotic patients. A trial to assess OP efficacy in lowering plasma ammonia levels versus placebo in cirrhotic patients after an upper gastrointestinal bleeding was performed. The primary outcome was a decrease in venous plasma ammonia at 24 hours.
Methods:
A total of 38 consecutive cirrhotic patients were enrolled within 24 hours of an upper gastrointestinal bleed. Patients were randomized (1:1) to receive OP (10 g/day) or glucosaline for 5 days.
Results:
The primary outcome was not achieved. A progressive decrease in ammonia was observed in both groups, being slightly greater in the OP group, with significant differences only at 120 hours. The subanalysis according to Child–Pugh score showed a statistically significant ammonia decrease in Child–Pugh C-treated patients at 36 hours, as well as in the time-normalized area under the curve (TN-AUC) 0–120 hours in the OP group [40.16 μmol/l (37.7–42.6); median (interquartile range) (IQR)] versus placebo group [65.5 μmol/l (54–126);p = 0.036]. A decrease in plasma glutamine levels was observed in the treated group compared with the placebo group, and was associated with the appearance of phenylacetylglutamine in urine. Adverse-event frequency was similar in both groups. No differences in hepatic encephalopathy incidence were observed.
Conclusions:
OP failed to significantly decrease plasma ammonia at the given doses (10 g/day). Higher doses of OP might be required in Child–Pugh A and B patients. OP appeared well tolerated.
Keywords
Introduction
Hepatic encephalopathy (HE) is a major complication of cirrhosis associated with high mortality and poor quality of life [Ferenci et al. 2002]; it also carries a high economic and social burden [Patel et al. 2012; Stepanova et al. 2012]. Multiple risk factors have been classically associated with the development of HE, including upper gastrointestinal bleeding (UGIB). UGIB induces a status of hyperammonemia through the intraluminal digestion of blood-nitrogenous compounds by colonic bacteria. In addition, the catabolic status caused by gastrointestinal bleeding provokes an increase in ammoniagenesis in different tissues [Olde Damink et al. 2003]. Ammonia and its transformation to glutamine in the astrocyte appear to be a key factor in HE development and pathogenesis [Albrecht and Norenberg, 2006; Butterworth, 2003; Shawcross et al. 2005]. Current strategies for the treatment of HE have been focused on lowering ammonia production through the administration of nonabsorbable disaccharides (NADs) or antibiotics. These therapies have been proved to be effective in HE primary and secondary prophylaxis [Agrawal et al. 2012; Bass et al. 2010; Sharma et al. 2012], but no effects on overt HE episodes or in survival have been shown [Als-Nielsen et al. 2004; Bass et al. 2010], except for rifaximin, with promising results in overt HE, but with a short period of follow up [Sharma et al. 2013]. Other strategies are based on promoting ammonia elimination; glycerol phenylbutyrate has proved its efficacy in secondary prophylaxis, but it has not been tested in cirrhotic-decompensated patients [Rockey et al. 2014]. Ornithine phenylacetate (OP) is a new drug proposed as an ammonia scavenger. This therapy is based on the capacity of ornithine to stimulate the activity of glutamine synthetase, hence incorporating ammonia into a ‘nontoxic’ molecule. The newly formed glutamine will combine with phenylacetate (PA), allowing the elimination of ammonia in the urine by its conversion into phenylacetylglutamine (PAGN). This strategy prevents the degradation of glutamine by intestinal glutaminase and avoids new formation of ammonia [Jalan et al. 2007]. OP has been proven to be effective in animal models [Oria et al. 2012] and well tolerated in cirrhotic-decompensated patients [Ventura-Cots et al. 2013]. OP has been evaluated in a phase I study conducted in healthy volunteers, as well as in a phase IIa study in patients with stable hepatic cirrhosis. The results confirmed that OP was well tolerated and had linear pharmacokinetics at doses up to 10 g infused over 4–24 hours [Hassanein et al. 2012]. Although our prior data supported the concept that this molecule might be effective in lowering ammonia [Ventura-Cots et al. 2013], no randomized clinical trial compared with placebo had been performed. The goal of the current study was to confirm the efficacy of OP in lowering plasma ammonia in patients with cirrhosis and UGIB, a relevant clinical scenario associated with high mortality and important complications [de Franchis, 2010; Garcia-Tsao and Bosch, 2010].
Methods
The study was a randomized, double-blind, placebo-controlled, multicenter study comparing the efficacy of OP versus placebo in cirrhotic patients with UGIB. Thirty-eight consecutive patients recruited from September 2012 to August 2014 admitted to the Hospital Vall d’Hebron and Hospital de la Santa Creu i Sant Pau (both in Barcelona) were included. A block size of 10 was used to randomize patients; randomization was also stratified by the centers.
The Institutional Review Boards at the study site and the Spanish Agency of Medications and Sanitary Products approved the study according to the World Medical Association’s Declaration of Helsinki. Written informed consent was obtained from subjects prior to enrolment. This study is a registered trial [ClinicalTrials.gov identifier: NCT01434108; EudraCT number: 2009-017819-16].
Patient selection
Subjects were eligible for inclusion if they met the following criteria: age between 18 and 85 years, had a diagnosis of cirrhosis as defined by clinical, laboratory or radiological findings and experienced a UGIB that was active within 24 hours prior to inclusion.
Subjects were excluded from enrolment if they exhibited one of the following criteria: need of mechanical ventilation, terminal illness, infection by human immunodeficiency virus, neurological comorbidities that impaired mental status, suspected hypersensitivity or allergic reaction to the drug components, electrocardiogram with QTcF > 500ms (Bazzet formula), creatinine > 1.5 mg/dl or need for hemodialysis. Additional causes of exclusion were the use of the following drugs: penicillin, probenecid, haloperidol, valproic acid, and systemic corticosteroids (because of their potential interaction with the study drug) and rifaximin (per protocol). In premenopausal women, a negative result in pregnancy test was required and breastfeeding was forbidden.
Study protocol
Patients were admitted to a semi-intensive care unit (Bleeding Unit) for at least 48 hours, until clinical stability was established. After this period of time, patients were transferred to a general-medicine ward. The standard protocol for the management of UGIB included: intravenous fluids (glucose and saline), perfusion of somatostatin (6 mg/24 hours), use of broad-spectrum antibiotics (ceftriaxone 1 g/daily, intravenously), omeprazole (20 mg/12 hours), nasogastric aspiration of blood and lactulose (initially as enema/12 hours; later orally, adjusted to 2–3 bowel movements/day). The use of rifaximin was prohibited as per protocol. Upper gastrointestinal endoscopy was performed during the first 24 hours and therapy was administered according to findings.
Study medication was prepared in 200 ml empty flasks (10 g/50 ml vials of OP diluted in 150 ml vials of sterile water for injection, or placebo, 200 ml glucose–saline solution) by the pharmacy department and blindly delivered to the study investigators. Medication was administered as an intravenous-continuous infusion, at 0.416 g/h for 5 days (10 g/day), provided the patients did not develop a stopping or withdrawal criteria. In addition, 1 day was required to carry out a clinical and biochemical assessment, and finally, a follow up at 28 days was performed.
Blood samples (10 ml) were collected every 12 hours during the first 48 hours and daily thereafter (up to day 6). A venous access was chosen to avoid discomfort to patients and because venous ammonia shows a good correlation with arterial ammonia [Ong et al. 2003]. Urinary samples were collected every 12 hours during the first 48 hours and daily thereafter (up to day 6). Complete urine collection was obtained via urinary catheter during the first 48 hours and by supervised 24-hour urine collection after catheter removal.
A follow up at day 28 was performed. Blood samples were centrifuged within 15 minutes of sampling and stored frozen at −80ºC.
Outcomes
The primary outcome was a decrease in average venous plasma ammonia at 24 hours. Secondary outcomes included: changes in plasma amino acids and ammonia between both groups at any timepoint; pharmacokinetic profiles of PA and ornithine in plasma, and PAGN in urine in order to explore mechanistic processes; HE [presence, duration and severity measured by the West Haven and Clinical Hepatic Encephalopathy Staging Scale (CHESS) scales]; bacterial infections during treatment period; and safety and tolerability of OP.
Analytical procedures
Ammonia was collected in refrigerated tubs, stored in ice until centrifugation (15 minutes, 3000 rpm, 4ºC), and frozen at −80ºC; the whole process was always performed in less than 15 minutes. Ammonia and amino-acid measurements were centralized in hospital Vall d’Hebron and performed in the Laboratory of Metabolic Diseases. Standard laboratory tests included: hemogram, reticulocyte count, prothrombine time and bilirubin, alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, albumin, sodium, potassium, creatinine and calcium plasma concentration.
Ammonia was measured in a Cobas 6000 analyzer (Roche Diagnostics Indianapolis, IN, USA) by standard methodology. Physiologic-free amino acids were analyzed as aminoquinoline derivatives by reverse-phase ultra-performance liquid chromatography (UPLC), Acquity-UPLC (AccQ-Tag chemistry, MassTrak AAA method and instruments, Waters, Milford, MA, US), using a binary solvent gradient at 42.5ºC and spectrophotometric detection at 260 nm following manufacturer instructions. Prior to derivatization and chromatography, an internal standard (norvaline) was added and samples were deproteinized by ultrafiltration [Narayan et al. 2011]. PA and PAGN were analyzed by liquid chromatography–tandem mass spectrometry according to the protocol set out by Laryea and colleagues [Laryea et al. 2010], with some modifications. PA and PAGN were analyzed by liquid chromatography–tandem mass spectrometry. (a) Reagents: deuterium-labeled internal-standard [N-(phenyl-d5-acetyl)-L-glutamine] (d5-PAG) (CDN isotopes, Pointe Claire, QB, Canada). (b) Sample preparation. (1) Standards: the calibration curve was prepared by diluting stock solutions of each standard with MiliQ water to cover a 0–12.92 µmol of PA and 0–32.30 µmol of PAGN concentration range, adding d5-PAGN as internal standard (until 3.5 µmol final), in conical plastic centrifuge tubes. (2) Plasma: 100 µl diluted 1:5 plasma was mixed with d5-PAGN (as in standards) and was ultrafiltered (10,000g × 30 minutes). (3) Urine: 100 µl 0.22 µmol filtered urine was diluted 1:200 with MiliQ water and mixed with d5-PAGN (as in standards). (c) Liquid chromatography–tandem mass spectrometry: instrumentation consisted of an Acquity-UPLC separation module (Waters, Milford, MA, USA) coupled to a Xevo TQ mass-spectrometry system with the Mass Lynx software (Waters, Manchester, UK). The analytical column was an Acquity-UPLC HSS C18 (2.1 mm × 100 mm). Sample elution was achieved by a 3-minute mobile-phase linear gradient consisting of an initial 40% solvent A (methanol) and 60% solvent B (1% formic acid in water), changing to 80% solvent A and 20% B from 0–2 minutes, back to 40% solvent A/60% B from 2–2.1 minutes, and equilibrating with these conditions from 2.1–3 minutes. Some 5 µl supernatant samples were injected into the system and the column temperature was maintained at 40ºC. Conditions for the mass-spectrometry analysis were: the ionization mode was a negative electrospray (ESI−); capillary voltage was 3kV; desolvation gas, heated to 450ºC and delivered at a flow rate of 1100 l/h; and cone-gas flow (argon) was 0.15 ml/min. Multiple reaction-monitoring transitions were optimized at the following conditions: dwell time: 0.073 seconds, PAGN: precursor ion 263 (m/z), product ion 145 (m/z), cone voltage 30 V, collision energy 15 eV; PA: precursor ion 135 (m/z), product ion 91 (m/z), cone voltage 20 V, collision energy 10 eV; d5-PAGN: precursor ion 268 (m/z), product ion 145 (m/z), cone voltage 30 V, collision energy 20 eV.
Clinical evaluation
Patients were evaluated at the time of initial infusion, every 12 hours during the first 2 days and every day thereafter until day 6; a follow up visit was performed 1 month later. The clinical exam included a 12-lead electrocardiogram, any complications related to cirrhosis, vital signs, and the presence of bacterial infections. The presence of HE was evaluated throughout the study with West-Haven criteria [Conn et al. 1977] and the CHESS [Ortiz et al. 2007].
Data analysis
The total sample size of 38 patients (17 patients per group plus 10% of losses) was calculated to detect a difference of 25 ± 25 standard deviation (SD) μmol/l in the mean ammonia average between the two groups at 24 hours (80% power using a one-sided test with alpha = 0.05).
The clinical trial data were independently monitored by the Academic Research Organization of the Vall d’Hebron Institute of Research. A statistical analysis plan was defined between the statistician of the Unit of Statistics and Bioinformatics of Vall d’Hebron Institute of Research and the study investigators before the conclusion of the study. A data-blind review was conducted between the monitors, the statistician and the study investigators before locking the trial database. The statistical analysis was carried out with Stata 13.1. Descriptive statistics were used to summarize the data. Quantitative variables were described as mean ± standard deviation for normally distributed variables or median and interquartile range if normality criteria were not met. Normality was assessed by Shapiro-Wilk test and quantitative variables were compared using the one-sided Wilcoxon Rank Sum Test. Percentages were calculated for categorical data. Qualitative variables were described as frequency and percentage, and the one-sided Fisher’s exact test was used for calculation. The Wilcoxon Rank Sum Test was used to compare the duration and severity of HE. Ammonia profile and changes were calculated with time-normalized area under the curve through 120 hours (TN-AUC0–120h). Subject’s regression slopes and group comparisons were made using a one-sided Wilcoxon Rank Sum test.
Results
Characteristics of patients
A total of 110 patients were assessed for eligibility; 72 were excluded for different reasons. The main reason for not participating was age, followed by a creatinine level higher than 1.5 mg/dl. Two patients had to be withdrawn from the study, one of them on day 5 because of a severe adverse event and the other because of a rebleeding during the first 12 hours (Figure 1).

Flow chart of patient selection.
Most of the patients in both groups were males, with a mean age of 55 years. Groups differed in cirrhosis etiology (70% alcohol in the OP group versus 44.4% in the placebo group, and 5.5% of hepatitis C virus in the OP versus 33.3% in the placebo group). Most of the patients in both groups belonged to Child–Pugh class B, with similar Model for End-stage Liver Disease (MELD) scores. All patients in both groups had presented with previous liver decompensation, being the most common UGIB, followed by ascites. Only one patient in the placebo group and two in the OP group had previously developed overt HE. At inclusion, ascites was the most frequent decompensation event (30% in the OP group versus 22.2% in placebo group), followed by HE; no bacterial infections were observed (Table 1). Hematological and biochemical parameters were in accordance of what is expected during a UGIB and no differences between groups were seen at baseline.
Characteristics of patients treated with ornithyne phenylactetate and placebo.

OP, ornithyne phenylactetate; HCV, hepatitis C virus; HE, hepatic encephalopathy; SBP, spontaneous bacterial peritonitis; SD, standard deviation; UGIB, upper gastrointestinal bleeding; Hb, hemoglobin; INR, international normalized ratio; ALT, Alanine transaminase; AST, Aspartate transaminase; Na, sodium; ECG, electrocardiogram; QTc corrected QT interval. Differences were not significant in any parameter except for HCV etiology; p < 0.05.
Primary outcome
The primary outcome was not achieved. OP failed to significantly decrease plasma ammonia at the given doses. A progressive decrease in ammonia was observed in both groups, being slightly greater in the OP group, with significant differences only at 120 hours (Figure 2). Although the decrease in ammonia at 24 hours in the OP group [−20.4 µmol/l (−39.20 to −2.80); median (IQR)] was almost twice that observed in the placebo group [−11.88 µmol/l (−36.75 to −26.35)], differences did not reach statistical significance. The subanalysis by Child–Pugh class showed a statistically significant ammonia decrease in Child C-treated patients at 36 hours (p = 0.018), as well as in the TN-AUC0–120h in the OP group [40.16 μmol/l (37.7−42.6)] versus placebo group [65.5 μmol/l (54−126); p = 0.024] (Table 2). However, the number of patients in this subgroup analysis was very small.
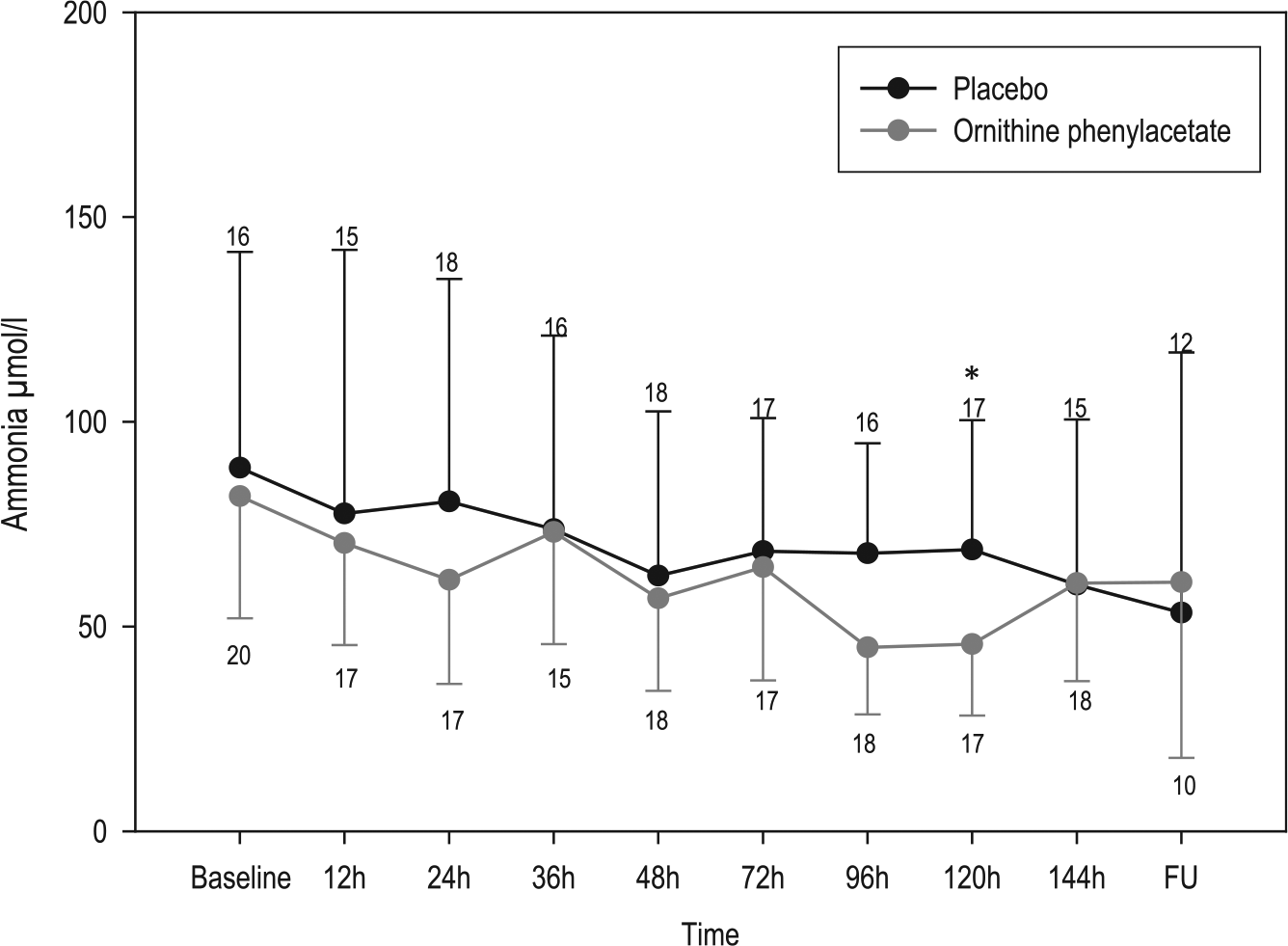
Plasma ammonia-concentration evolution during the study period in the ornithine phenylacetate and placebo groups.
Ammonia levels expressed as time normalized area under the curve from 0 to 120 hours (TN-AUC0–120h) of treated and control patients distributed by Child–Pugh score.

OP; Ornithine phenylacetate, IQR; interquartile range.
Other analytical parameters
A progressive decrease in glycine and glutamine was observed in the treated group as compared with the placebo group, reaching a peak at day 5 in both cases. Differences between both groups started at the 24-hour point and were maintained thereafter (Figure 3). Plasma PA levels differed between groups during the entire study (Table 3), the TN-AUC0–120h in the placebo group was 1.6 μmol/l (0.7−3.3) versus 111.6 μmol/l (63.5−148.5) in the OP group (p < 0.05). Once the 36-hour dose was reached, PA levels in the OP group remained stable until the end of infusion, never achieving the toxic values that have been reported at a much higher range (approximately 7000 μmol/l) [Thibault et al. 1994]. Urinary levels of PAGN, the main elimination pathway of OP, showed remarkable differences between both groups (Table 3). Total amount of PA excretion accounted for 87% of PA administered as OP at the end of the study.

Glutamine (A) and glycine (B) plasma levels in treated and placebo patients.
Levels of phenylacetate in plasma, accumulated phenylacetylglutamine in urine (PAGNU), PAGNU adjusted to creatinine and percentage of PA eliminated in urine in patients treated with ornithine phenylacetate and placebo.

OP, ornithyne phenylactetate; PA, phenylacetate; PAP, phenylacetate in plasma; PAGNU, accumulated phenylacetylglutamine in urine; CreatinineU, creatinine in urine; SD, standard deviation.
p < 0.05, OP group compared with placebo.
In OP group only.
Molar ratios were calculated by dividing PAGNU accumulated (mmol) by OP administered (mmol), considering that 1 mol of PAGN results from 1 mol of PA (or OP).
Clinical outcome
Per standard of care, all patients underwent an upper gastrointestinal endoscopy during the first 24 hours of admission. The main cause for UGIB in both groups was esophageal variceal bleeding (45% OP versus 83.3% placebo; p = 0.1); all of these patients underwent drug treatment and variceal ligation. In the OP group, duodenal or gastric ulcers were frequent etiologies and suitable endoscopic therapies were performed. Three patients in the placebo group and seven in the OP group did not require endoscopic treatment. Only one patient in the OP group presented with rebleeding during the first 24 hours and underwent a transjugular intrahepatic portosystemic shunt. Nine patients in the placebo group and 12 in the OP group required blood transfusion; no difference between the total number of blood units transfused and the initial hemoglobin levels were observed between both groups (Table 1). The exposure to blood in the digestive tract was similar for both groups, since the ratio of isoleucine/(leucine plus valine), a good parameter for estimation of the amount of blood ingested, showed no differences between groups (Supplementary Figure 1) [Dejong et al. 1996; Olde Damink et al. 1997].
Ascites was the most frequent decompensation event at inclusion: four patients in the placebo group and six in the OP group. Interestingly, three patients in the placebo group and none in the OP group developed ascites during the study.
Three patients in each group exhibited HE at inclusion. The median time of resolution was 24 hours in both groups. One patient in the OP group presented with HE grade III, and two with grade II, while in the placebo group, all patients had HE grade II. During infusion time, two patients in the placebo group (grade II and grade III) and one in the OP group developed HE (grade I). Lactulose exposure during the study was 90% in the placebo group and 95% in the OP group.
Only one patient in the placebo group presented with a bacterial infection (pneumonia because of bronchial aspiration, secondary to the endoscopic procedure).
Safety and tolerability
The incidence of adverse events reported during the study was similar between the OP group (44.72%) and the placebo group (55.28%), as they were the more common adverse events (Table 4). Only one adverse event (nausea) in the OP group was considered related to medication. Ten severe adverse events were reported during the study: four in the placebo group and six in the OP group. Only three of these events were reported during drug infusion: one in the OP group and two in the placebo group (Table 5).
Most frequent adverse events and severe adverse events in the treated (ornithine phenylacetate) and placebo group during treatment infusion (5 first days) and follow-up period (FU, 28 days).
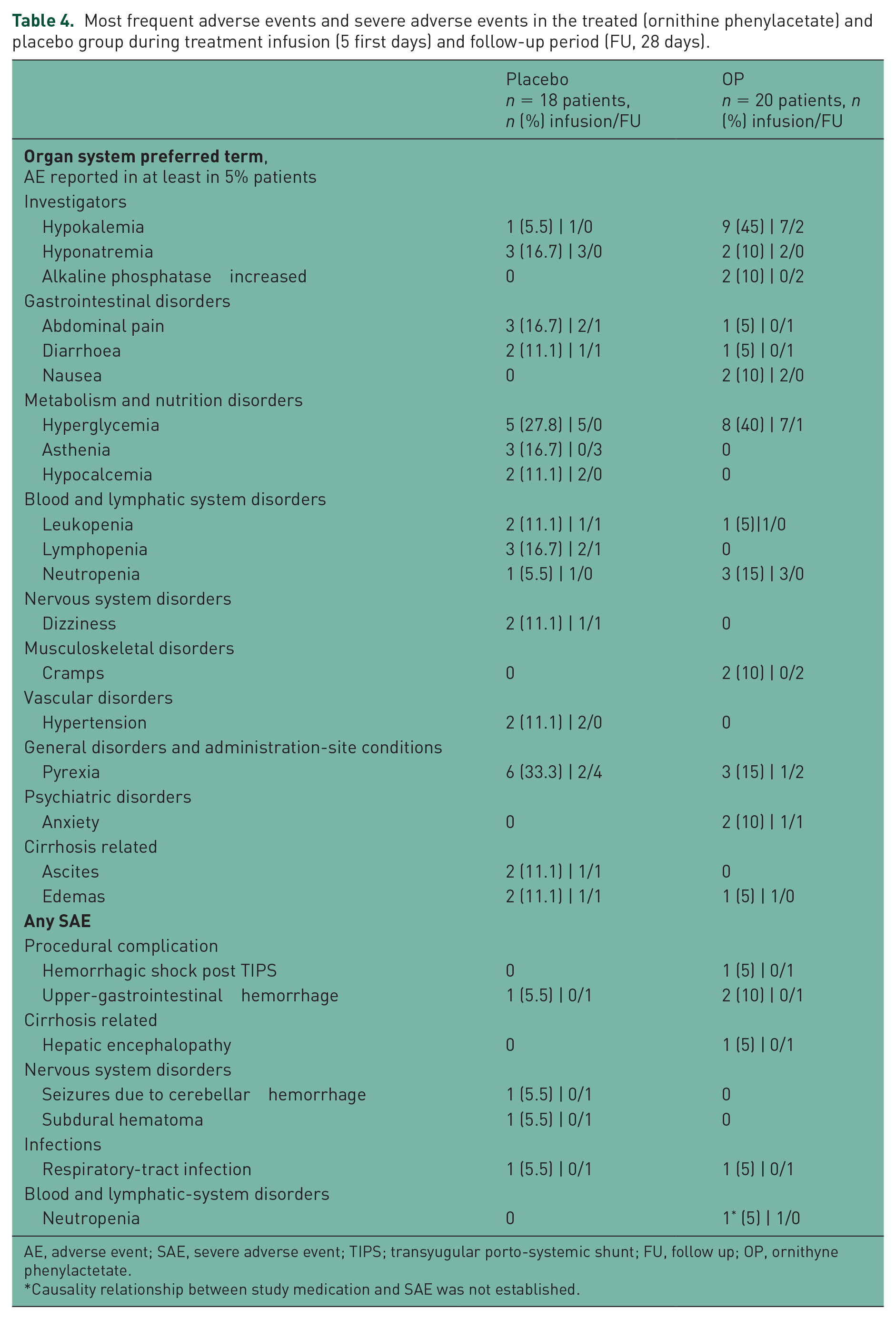
AE, adverse event; SAE, severe adverse event; TIPS; transyugular porto-systemic shunt; FU, follow up; OP, ornithyne phenylactetate.
Causality relationship between study medication and SAE was not established.
Total number of adverse events with grading and presentation time in treated (ornithine phenylacetate) and placebo patients.
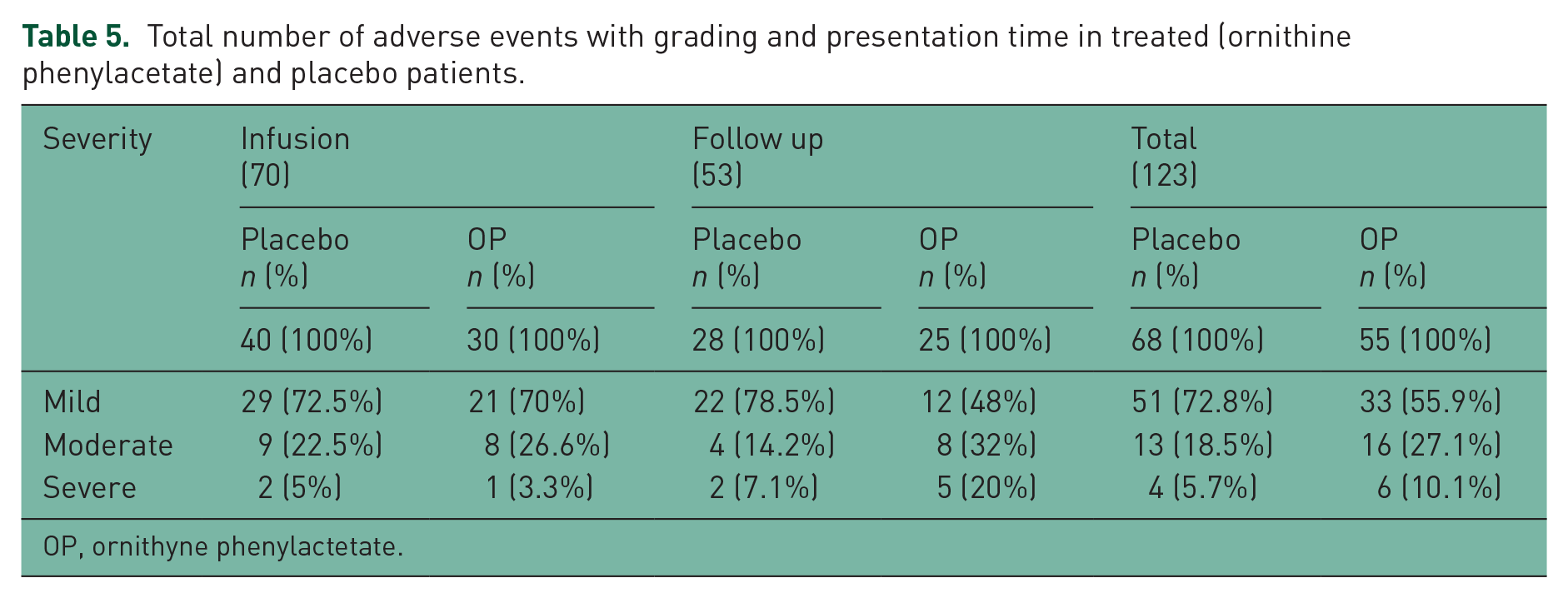
OP, ornithyne phenylactetate.
Discussion
Based on the results of the present study, OP was not able to obtain a significant decrease in ammonia levels at 24 hours of therapy compared with placebo, and consequently failed to achieve the primary endpoint.
OP has proven safe and well tolerated in cirrhotic-decompensated patients [Ventura-Cots et al. 2013] and preclinical studies have shown its potential effect in lowering plasma ammonia [Oria et al. 2012]. Although the trial was not successful in reaching this goal, mean ammonia levels were lower at every timepoint in the OP group, with statistically significant differences at 120 hours. In addition, the TN-AUC0–120h was significantly lower in the Child–Pugh C patients of the OP group compared with the placebo group, showing differences of 13.4µmol/l between groups. Differences of this magnitude been previously described as clinically significant (has been proven effective in decreasing the incidence of HE) in HE secondary-prophylaxis studies [Agrawal et al. 2012; Rockey et al. 2014]. However, the data obtained in Child–Pugh C patients should not be considered definitive, since the total number of Child–Pugh C patients included was very low.
Similarly to our prior safety and tolerability study [Ventura-Cots et al. 2013], only patients with baseline plasma ammonia above the upper normal limit (⩾50 µmol/l) seem to obtain benefit from ammonia-scavenging drugs. It is important to consider the great individual variability in ammonia levels, not only in the OP group, but also in the placebo group. This variability has been previously described in healthy subjects, patients with urea-cycle disorders and patients with hepatic disease [Mokhtarani et al. 2013], suggesting not only the relevance of sample processing, but the participation of genetic factors, such as the variability in the glutaminase gene [Romero-Gomez et al. 2010].
It is important to emphasize that the mean plasma concentration of PA achieved in our treated patients [maximum plasma PA level of 24.7 µg/ml at 36 hours of infusion (181 µmol/l), Table 3] is much lower than the concentrations obtained with similar therapies, such as glycerol phenylbutyrate (Ravicti ®) (Cmin: 84.4 µg/ml–Cmax: 292 µg/ml) [Ghabril et al. 2013; Hyperion Therapeutics, 2014] or sodium phenylacetate (Ammonul ®), (Cmin: 120 µg/ml–Cmax: 4652 µg/ml) [Food and Drug Administration, 2005]. We hypothesize that higher doses of OP, ranging from 15 to 20 g daily, would obtain higher plasma PA levels and therefore achieve a higher drop in plasma ammonia, to levels that have been historically associated with a low incidence of HE. These higher doses of OP should be probably applied to Child–Pugh A and B patients, since in our subgroup analysis, OP appeared to have a higher effect in Child–Pugh C patients with the current dose of 10 g/day. In addition, data from other studies indicate that Child–Pugh C patients have higher exposure to PA than Child–Pugh A and B patients [Ghabril et al. 2013; McGuire et al. 2010; Hyperion Therapeutics, 2014]; the efficacy of higher doses of OP should be confirmed in future studies.
The total number of patients included in our study (38 patients) was calculated to detect a difference of 25 ± 25 (SD) μmol/l in the mean ammonia average between the two groups at 24 hours. This calculation was based in the results of our pilot study [Ventura-Cots et al. 2013], where the decrease of plasma ammonia during the first 24 hours in the treated group was compared with a historical cohort of 17 patients who exhibited a steady state of plasma ammonia during the first 24 hours; this steady state was not observed in the placebo group of the present study. This fact has been critical to determine the lack of power in the present study to achieve the main objective.
The decrease of plasma glutamine concentration during time, as well as the increased in PAGN detected in urine, confirms the proposed mechanism of action (combining glutamine to PA and enhancing urinary excretion). OP is a new drug that acts by providing an alternate pathway for ammonia removal in the form of urinary PAGN [Jalan et al. 2007]. In our study, the recovery of as much as 87% PA administered as urinary PAGN indicates that most of the drug is metabolized and participates in ammonia removal. The plasma amino-acid profile of the OP-group patients also seems to support the existence of other ammonia-removal pathways promoted by OP and previously described in preclinical studies, such as the combination with glycine and its consequent decrease in plasma, followed by their elimination as phenylacetylglycine in urine [Kristiansen et al. 2014]. This hypothesis has not been confirmed in our study, since the urinary determination of phenylacetylglycine was not carried out.
Our trial had a similar incidence of HE episodes between both groups at baseline, during infusion and during follow up. Taking into account the incidence of HE during cirrhosis decompensation (16–21%) [EASL, 2010] and the sample size of our study (n = 38), differences in the incidence of HE were not expected. We did not observe significant differences in baseline and follow-up ammonia levels in patients with HE, treated with OP or placebo.
The study confirms that OP, at the present dose, is a well-tolerated drug, even in decompensated cirrhotic patients. The incidence of adverse events and severe adverse events was the same in both groups, and it is important to underline that most of the adverse events recorded in both groups developed during the follow-up period, after drug infusion.
OP appears to be a well tolerated and promising drug for the treatment of ammonia-related complications of cirrhosis, even in patients with poor liver function. The current study did not achieve the primary objective, but this was probably due to individual variability, a small number of patients included and mainly, too low a dose of OP administered. On the other hand, the consistent decrease in ammonia levels during therapy in the treated group and the safety profile of OP indicate that with higher doses, especially in Child–Pugh A and B patients, a clinical effect will probably be achieved.
Footnotes
Funding
This study was funded in part by the Spanish Ministry of Health, grant number, FIS 10/1028.
This study was funded in part by the Instituto de Salud Carlos III, grant number, TRA-190.
Macarena Simón-Talero is a recipient of a ‘Río Hortega’ fellowship grant from the ISCIII.
Conflict of Interest statement
Potential competing interests: Juan Córdoba (deceased) received consulting fees from Ocera Therapeutics Inc. The remaining authors declare that they have nothing to disclose.
Study medication was purchased from Ocera Therapeutics Inc., by the promoters of the study.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.