
Abstract
Pre-eclampsia (PE) is the most frequently encountered medical complication during pregnancy. It is characterized by a rise in systemic vascular resistance with a relatively low cardiac output and hypovolemia, combined with severe proteinuria. Despite the hypovolemia, renin–angiotensin system (RAS) activity is suppressed and aldosterone levels are decreased to the same degree as renin. This suggests that the RAS is not the cause of the hypertension in PE, but rather that its suppression is the consequence of the rise in blood pressure. Abnormal placentation early in pregnancy is widely assumed to be an important initial event in the onset of PE. Eventually, this results in the release of anti-angiogenic factors [in particular, soluble Fms-like tyrosine kinase-1 (sFlt-1)] and cytokines, leading to generalized vascular dysfunction. Elevated sFlt-1 levels bind and inactivate vascular endothelial growth factor (VEGF). Of interest, VEGF inhibition with drugs like sunitinib, applied in cancer patients, results in a PE-like syndrome, characterized by hypertension, proteinuria and renal toxicity. Both in cancer patients treated with sunitinib and in pregnant women with PE, significant rises in endothelin-1 occur. Multiple regression analysis revealed that endothelin-1 is an independent determinant of the hypertension and proteinuria in PE, and additionally a renin suppressor. Moreover, studies in animal models representative of PE, have shown that endothelin receptor blockers prevent the development of this disease. Similarly, endothelin receptor blockers are protective during sunitinib treatment. Taken together, activation of the endothelin system emerges as an important pathway causing the clinical manifestations of PE. This paper critically addresses this concept, taking into consideration both clinical and preclinical data, and simultaneously discusses the therapeutic consequences of this observation.
Keywords
Introduction
Pre-eclampsia (PE) is the most frequently encountered medical complication during pregnancy. PE is not simply de novo onset of hypertension and proteinuria in the last half of pregnancy, but rather a syndrome involving many organs, of which the clinical spectrum ranges from relatively mild to life-threatening [Steegers et al. 2010]. Currently, treatment of PE consists of treating the elevated blood pressure and prevention of seizures, but the ultimate remedy is delivery of the placenta, indicating that the placenta is a central culprit in the pathogenesis of PE [Steegers et al. 2010].
The etiology of PE is unknown. A large body of evidence, supported by preclinical models of PE, indicates that abnormal placentation early in pregnancy is an important initial event in the onset of PE [Roberts and Redman, 1993; Myatt, 2002]. This abnormal placentation stimulates the production of anti-angiogenic factors and cytokines, resulting in generalized vascular dysfunction and the clinical manifestations of PE. In the past several years, activation of the endothelin (ET) system has emerged as an important pathway causing the clinical manifestations of PE [Makris et al. 2007; Maynard et al. 2008; Verdonk et al. 2014]. This paper critically addresses this concept, taking into consideration both clinical and preclinical data.
Pathogenesis and manifestations of pre-eclampsia
Clinically, PE is divided into 2 types: early-onset PE before 34 weeks of gestation and late-onset PE at, or after 34 weeks of gestation [Von Dadelszen et al. 2003]. The incidence of PE is 3–8% worldwide. The pathogenesis of PE involves two stages [Redman, 1992; Redman et al. 2005; Seki, 2014]. In stage one, aberrant shallow cytotrophoblast invasion in the maternal spiral arteries supplying the placenta results in poor placentation [Brosens, 1964]. This poor placentation is postulated as the root cause of stage two, consisting of repeated periods of placental hypoxia and reperfusion injury, resulting in oxidative stress and an increased production of placental factors such as soluble Fms-like tyrosine kinase 1 (sFlt-1), soluble endoglin, agonistic auto-antibodies to the angiotensin (Ang) II type 1 receptor (AT1R-AA) and inflammatory cytokines [Maynard et al. 2008; Naljayan et al. 2013; Seki, 2014]. In the maternal circulation these factors cause activation of endothelial cells and generalized endothelial dysfunction, leading to the clinical manifestations of PE. PE is a multifaceted disorder: in addition to hypertension and proteinuria, it can also affect the central nervous system, lungs, liver, and the heart [Steegers et al. 2010]. PE may increase the risk of eclampsia and the development of the HELLP (hemolysis, elevated liver enzymes and low platelets) syndrome, a severe condition characterized by disseminated intravascular coagulation, acute renal failure and pulmonary edema that can end in maternal death [Haram et al. 2009]. PE is cured by delivery of the placenta, which to date is the only effective treatment of PE. Although beneficial for the mother, preterm delivery may compromise the health of the infant both acutely and chronically; hence treatments to prevent or alleviate PE in order to prolong pregnancy are urgently needed [Friedman et al. 1999].
Hemodynamics and the renin–angiotensin system in pre-eclampsia
Compared with normal pregnancy, PE is characterized by a rise in systemic vascular resistance with a relatively low cardiac output and hypovolemia [Hall et al. 2011]. This rise in systemic vascular resistance is accompanied by suppression of the renin–angiotensin system (RAS) [Powe et al. 2011]. The latter is somewhat unexpected in view of the reduced circulating volume. It might simply represent protection against a further rise in blood pressure, related to the fact that blood pressure itself inversely affects renin release. An alternative reason for the suppression of the RAS is the production of AT1R-AA by the placenta of PE patients [Verdonk et al. 2015]. Such antibodies, by activating the AT1 receptor, should indeed suppress renin release from the kidneys (the consequence of the so-called negative feedback loop between Ang II and renin release), but would also be expected to increase aldosterone synthesis in the adrenal gland. As a result, the aldosterone/renin ratio in PE should be higher as compared with the ratio in healthy pregnant women. Unexpectedly, this turned out not to be the case [Verdonk et al. 2015]. Moreover, AT1R-AA are not present in all PE cases and can even be detected in healthy pregnant women [Walther et al. 2005]. Therefore, despite preclinical studies supporting a role for AT1R-AA in the pathogenesis of PE [Faas et al. 1994; Zenclussen et al. 2004; Zhou et al. 2008], there is still doubt about the in vivo importance of AT1R-AA in PE.
It has been well established that the blood pressure rise to exogenous Ang II in PE is larger than in healthy pregnant women [Gant et al. 1973; Baker et al. 1992]. Obviously, the generalized endothelial dysfunction in PE may account for the increased Ang II sensitivity [Wenzel et al. 2011]. In addition, by investigating subcutaneous resistance vessels obtained from pregnant women with and without PE ex vivo, we observed that the increased vasoconstrictor response to Ang II in PE involves Ang II type 2 (AT2) receptors [Verdonk et al. 2015]. Normally, this receptor induces vasodilation, but often its phenotype changes under pathological conditions [Moltzer et al. 2010]. Possibly, therefore, the enhanced sensitivity to Ang II in PE is additionally due to an upregulation of constrictor AT2 receptors.
Recently, Gennari-Moser and colleagues proposed that vascular endothelial growth factor (VEGF) stimulates aldosterone production, both directly and indirectly, the latter by enhancing adrenal capillary density [Gennari-Moser et al. 2013]. PE patients display elevated levels of the VEGF-binding soluble receptor, sFlt-1, and thus would be expected to have suppressed aldosterone levels and a decreased aldosterone/renin ratio [Gennari-Moser et al. 2013]. However, as discussed above, although aldosterone levels are indeed diminished in PE, so are renin levels, and the aldosterone/renin ratio is unaltered [Verdonk et al. 2015]. This argues against VEGF being an important determinant of the suppressed aldosterone levels in PE. Rather, a factor that suppresses renin seems to be involved, which, consequently, would lower aldosterone to the same degree, simply because of RAS suppression. This factor could be the rise in blood pressure, as discussed above, although normally, the inverse relationship between blood pressure and renin release is rather modest [Danser et al. 1998].
Collectively, the findings summarized here indicate that RAS suppression is counterintuitive in PE, given the hypovolemia in this disease. It might be the consequence of the rise in blood pressure. If so, remaining questions are: what causes this rise in blood pressure, if not the RAS, and, whether blood pressure is truly the only determinant of the suppression of renin release in PE. As outlined below, emerging data indicate that activation of the ET system may be the reason.
The endothelin system
ETs are a family of three 21-amino-acid peptides (ET-1, ET-2 and ET-3), each encoded by distinct genes (EDN1, EDN2 and EDN3) [Yanagisawa, 1994; Yanagisawa et al. 1998a]. The EDN genes encode the prepro form of ETs (prepro-ETs). Prepro-ETs are cleaved at dibasic sites to big ETs by a furin-like endopeptidase [Pollock and Opgenorth, 1993]. Big ETs are biologically inactive. They undergo further modification by one of the ET-converting enzymes (ECEs) to yield the biologically active ETs (Figure 1) [Inoue et al. 1989; Takahashi et al. 1993; Xu et al. 1994; Yanagisawa et al. 1998b]. There are three isoforms of ECE (ECE-1, ECE-2 and ECE-3), localized in endothelial and smooth muscle cells, cardiomyocytes and macrophages [Xu et al. 1994; Maguire et al. 1997; Fukuchi and Giaid, 1998]. Of the ET family, ET-1 is the predominant member. It is synthesized and secreted by a range of cells, including endothelial cells and the syncytiotrophoblasts of the placenta [Rubanyi and Polokoff, 1994]. ET secretion occurs constitutively and upon activation from stores in the so-called Weibel–Palade bodies of endothelial cells [Malassine et al. 1993; Van Mourik et al. 2002]. Several stimuli like Ang II, norepinephrine, thrombin, cytokines, growth factors, hypoxia, insulin, shear stress, free radicals, but also ET-1 itself, have been reported to induce endothelial ET-1 release [Levin, 1995; Jougasaki et al. 2002; Romani De Wit et al. 2004; Marasciulo et al. 2006; Khimji and Rockey, 2010]. ET-1 is released towards the basolateral side of these cells, acting primarily as a paracrine or autocrine peptide [Wagner et al. 1992].

Endothelin (ET) synthesis and receptors in the vascular wall. ECE, endothelin-converting enzyme; ETA, ETB, endothelin type A and type B receptor. See text for explanation.
ETs elicit their effect by binding to the cell-membrane G-protein-coupled ET type A and B (ETA and ETB) receptors, mapped on chromosomes 4 and 13 [Levin, 1995]. These receptors can be differentiated pharmacologically based on their affinity for the ETs. The ETA receptor has a 10-fold greater affinity for ET-1 and ET-2 than for ET-3, whereas the ETB receptor has similar binding affinity for all ETs [Watanabe et al. 1989; Sakurai et al. 1990]. The ETA receptor binds ET-1 almost irreversibly [Hilal-Dandan et al. 1997]. Furthermore, cross-talk between ETA and ETB receptors has been reported, in a way that inhibition of one receptor subtype will free the other receptor subtype from the inhibition [Fukuroda et al. 1996].
ETA and ETB receptors are widely distributed in various tissues, including the lungs, kidneys, liver, heart, brain, heart, eye, ovaries and adrenal glands [MacCumber et al. 1989, 1990; Masaki, 2004]. The majority of the ETA receptors are located on vascular smooth muscle cells (VSMC) whereas the ETB receptors are located on endothelial cells, VSMC, and epithelial cells [Masaki et al. 1992; Seo and Lüscher, 1995; D’orleans-Juste et al. 2002; Motte et al. 2006]. Activation of ETA and ETB receptors on VSMCs initiates vasoconstriction and cell proliferation, whereas activation of the ETB receptor on endothelial cells mediates vasodilation by releasing nitric oxide (NO) and prostacyclin [Ekelund et al. 1994; Lankhorst et al. 2013]. Binding of these receptors to different G-proteins is the most likely explanation for these diverse effects. Many factors can affect ET receptor expression. Insulin is known to cause an increase of ETA receptors in VSMCs, whereas the ETB receptor in endothelial cells is upregulated by tumor necrosis factor-α and basic fibroblast growth factor 2 [Frank et al. 1993; Smith et al. 1998; Francis et al. 2004].
ET-1 is rapidly cleared from the circulation. Studies revealed a plasma half-life ranging from 1.4 to 3.6 min, while the vasoconstriction has been shown to persist up to several hours [Vierhapper et al. 1990; Weitzberg et al. 1991]. The lung is able to clear more than 40% of ET-1. The kidneys and liver also play a role in the clearance of ET-1 from the circulation [Gandhi et al. 1993]. ETB receptors are likely responsible for this clearance, because intravenous infusion of the ETB receptor antagonist BQ788 extensively inhibited ET-1 uptake by the lungs and kidneys and increased plasma ET-1 levels while the ETA receptor antagonist BQ123 had no such effects [Fukuroda et al. 1994; Dupuis et al. 1996].
Endothelin-1 in preclinical models of pre-eclampsia
In the past decade many animal models have been developed to replicate the various aspects of human PE. Here, we discuss the most important models.
Reduced uterine perfusion pressure
The reduced uterine perfusion pressure (RUPP) model in rats recapitulates many of the hallmarks of PE [Alexander et al. 2001a]. In this model, blood flow to the uterus is partially occluded at day 14 of gestation, resulting in placental ischemia [Alexander et al. 2001a; Granger et al. 2002]. Findings in this model demonstrate that blood pressure, proteinuria, renal expression of prepro-ET-1 in both medulla and cortex, and plasma ET-1 are significantly elevated, whereas renal function and NO production are impaired, as compared with pregnant controls [Alexander et al. 2001b; Kiprono et al. 2013]. This model is also associated with fetal growth restriction. Treating rats with an ETA receptor blocker abolished the rise in blood pressure and the renal dysfunction, demonstrating that the enhanced ET-1 production, via activation of the ETA receptor, mediates the hypertension and proteinuria in this model [Alexander et al. 2001b]. Aside from increased ETA receptor-mediated vasoconstriction, downregulation of the vasodilator microvascular ETB receptor may also contribute to the PE-like findings in this model [Mazzuca et al. 2014].
Soluble Fms-like tyrosine kinase-1 elevation
An imbalance between pro- and anti-angiogenic factors, especially an increase in sFlt-1, is an important initial event in the pathogenesis of PE [Fiore et al. 2005; Maynard et al. 2008]. This has stimulated the development of rat models in which sFlt-1 has been increased in various ways [Maynard et al. 2003; Murphy et al. 2010, 2012]. Maynard and colleagues injected the tail vein with recombinant adenovirus encoding the murine sFlt-1 gene [Maynard et al. 2003], while Murphy and colleagues infused sFlt-1 at a rate of 3.7 µg/kg/day for 6 days [Murphy et al. 2010]. A three-fold increase in plasma sFlt-1 was reached in both models. The rats developed significant hypertension, proteinuria and glomerular endotheliosis [Maynard et al. 2003]. Moreover, increased plasma ET-1 levels and an increased prepro-ET-1 mRNA expression in both kidney and placenta were observed [Murphy et al. 2010]. Treatment with an ETA receptor antagonist abolished the hypertensive responses in both studies, while having no effect in normotensive rats. This indicates that the rise in blood pressure was mediated by the ET system.
Injection of angiotensin II type 1 receptor agonistic auto-antibodies
Zhou and colleagues injected pregnant mice with 800 µg IgG isolated from sera of either normotensive pregnant women or PE patients, the latter containing AT1R-AA [Zhou et al. 2008]. Mice injected with IgG from PE patients demonstrated hypertension, proteinuria, glomerular endotheliosis, elevated renal prepro-ET-1, placental abnormalities and smaller fetuses. Moreover, their sFlt-1 concentration was also significantly increased, in contrast to the sFlt-1 level in nonpregnant mice injected with IgG from either normotensive pregnant women or PE patients, which remained very low because the major source of sFlt-1 (the placenta) was missing [Herse et al. 2007]. Notably, pregnant mice injected with IgG isolated from normotensive pregnant women did not develop the above symptoms. Co-injecting the PE IgG-exposed mice with BQ123, an ETA receptor blocker, abolished these features, again indicating the involvement of ET-1 production in the pathophysiology of PE [Zhou et al. 2011]. Identical observations were made in pregnant rats: infusion of rat AT1R-AA resulted in hypertension and ET-1 upregulation in kidney and placenta, and treatment with the ETA receptor antagonist ABT-627 prevented the blood pressure rise [LaMarca et al. 2009]. This leaves the question: how do AT1R-AA activate the ET system? A link between AT1 receptor activation and ET-1 release was already noted more than 20 years ago [Dohi et al. 1992]. Zhou and colleagues making use of human placental villous explants, recently showed that this involved the tumor necrosis factor-α/interleukin-6 signaling pathway, since antibodies against these cytokines or their receptors prevented the AT1R-AA-induced ET-1 secretion [Zhou et al. 2011].
Altered anti-angiogenic state and endothelin-1 overexpression
Anti-angiogenic treatment in patients with cancer with either antibodies to VEGF or inhibitors of the VEGF receptors, so-called receptor tyrosine kinase inhibitors (RTKI), induces hypertension and renal toxicity [Hayman et al. 2012]. In patients treated with the RTKI sunitinib, we were the first to show that the rise in blood pressure was associated with increased circulating ET-1 levels [Kappers et al. 2010]. Administration of sunitinib to rats for 8 days also induced hypertension, proteinuria, renal function impairment, glomerular endotheliosis, as well as elevated circulating ET-1 levels [Kappers et al. 2011]. In fact, the renal histological changes observed during sunitinib exposure closely resembled those observed in PE. This is not too surprising, since VEGF inhibition (with an RTKI) will accomplish the same effect as VEGF inactivation or binding (with sFlt-1). Co-administration of the dual ETA/ETB receptor blocker macitentan could prevent the hypertension and proteinuria induced by sunitinib, indicating that, like in experimental models of PE, activation of the ET axis mediates the hypertension and proteinuria induced by anti-VEGF treatment. Taken together, these data indicate that RTKI treatment induces a PE-like syndrome involving ET-1, albeit in the absence of pregnancy.
Lastly, the consequences of endothelial ET-1 excess are further emphasized by a recent study by Rautureau and colleagues [Rautureau et al. 2015]. They developed a mouse model of inducible endothelium-specific ET-1 overexpression. Remarkably, such endothelial ET-1 overexpression led to hypertension, in an ETA receptor-dependent manner, but not to vascular or kidney injury, or changes in kidney perfusion or function. This suggests that renal damage, if occurring, depends on renal ET-1 overexpression rather than elevated circulating ET-1 levels. Our data in sunitinib-treated rats, showing that renal toxicity requires higher doses [Lankhorst et al. 2015], and that antihypertensive treatment with calcium antagonists, ACE inhibitors or macitentan differentially affects blood pressure and kidney damage [Lankhorst et al. 2014], fully confirms this view.
Endothelin-1 in clinical pre-eclampsia
Studies examining ET-1 in normal and PE pregnancies observed a two- to three-fold rise of circulating ET-1 in PE pregnancies compared with normal pregnancies (Table 1), with some studies indicating a positive correlation with the severity of the disease [Nova et al. 1991; Aydin et al. 2004; Baksu et al. 2005; Bernardi et al. 2008; Aggarwal et al. 2012; Karakus et al. in press; Verdonk et al. 2015]. In agreement with the latter, patients with the HELLP syndrome displayed even higher ET-1 levels than PE patients [Nova et al. 1991; Bussen et al. 1999; Karakus et al. in press]. Elevated ET-1 levels or expression in amniotic fluid and blood vessels obtained from PE patients versus healthy pregnant women [McMahon et al. 1993; Wolff et al. 1996; Faxen et al. 1997; Napolitano et al. 2000] confirm the concept that ET-1 levels are uniformly elevated in this disease. Importantly, multiple regression analysis revealed that ET-1 is not only an independent determinant of both the blood pressure rise and proteinuria in PE, but also a renin suppressor [Verdonk et al. 2015]. Animal studies support the latter [Ritthaler et al. 1995; Ortiz-Capisano, 2014].
Plasma ET-1 levels in healthy pregnant women, pre-eclampsia and HELLP.

HELLP, hemolysis, elevated liver enzymes and low platelet syndrome.
It remains to be determined what causes the rise in ET-1. The strong correlation between sFlt-1 and plasma ET-1 (Figure 2) [Aggarwal et al. 2012; Verdonk et al. 2015], as well as the observation that ET-1 rises dose-dependently in rats treated with the VEGF inhibitor sunitinib [Lankhorst et al. 2015], suggest that the rise in ET-1 is the direct consequence of VEGF inactivation or inhibition. Since ET-1 itself triggers oxidative stress in the placenta, which in turn may result in increased production of placental factors such as sFlt-1 [Fiore et al. 2005], a vicious circle seems to arise which steadily raises circulating ET-1 in PE, subsequently affecting blood pressure, kidney function and RAS activity (Figure 3).

Relationship between soluble Fms-like tyrosine kinase-1 (sFlt-1) and endothelin-1 (ET-1) in plasma obtained from healthy pregnant women and women with preeclampsia. Data have been modified from [Verdonk et al. 2015].
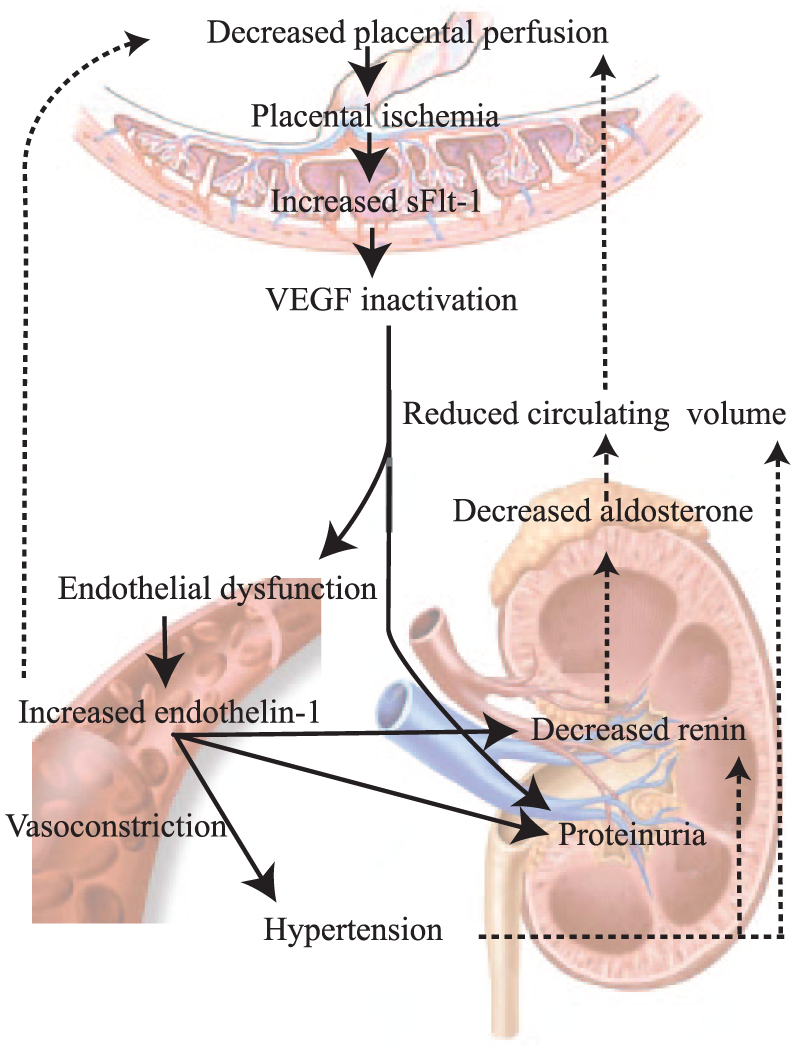
Unifying model depicting the central role of endothelin-1 (ET-1) in pre-eclampsia. Decreased perfusion of the placenta results in placental hypoxia and soluble Fms-like tyrosine kinase-1 (sFlt-1) release. SFlt-1 binds free vascular endothelial growth factor (VEGF), thereby inactivating this factor and inducing endothelial dysfunction. As a consequence, ET-1 production is turned on, which not only induces hypertension and proteinuria but also suppresses renin release. Such suppression will also occur due to the rise in blood pressure. The renin suppression is accompanied by a parallel aldosterone suppression, illustrating that the latter is entirely due to diminished angiotensin generation. Diminished renin–angiotensin-aldosterone system activity combined with high blood pressure results in a reduced circulating volume, thereby further decreasing placental perfusion. In addition, ET-1 induces sFlt-1 release from the placenta, thereby generating a deleterious feed-forward mechanism.
Therapeutic implications
NO, cleaved from L-arginine by NO synthase, is a well known suppressor and physiological antagonist of ET-1 [Boulanger and Lüscher, 1990; Brunner et al. 1995; Ohkita et al. 2002]. Indeed, sFlt-1-infused pregnant rats responded well to L-arginine administration: both the renal mRNA expression of ET-1 and maternal blood pressure decreased, while vascular function and fetal weight improved [Murphy et al. 2012]. A 3-week therapy of L-arginine also decreased blood pressure in women with PE, while prolonged treatment additionally improved fetal conditions and infant outcome [Rytlewski et al. 2005]. Facchinetti and colleagues confirmed the acute blood pressure-lowering effect of L-arginine in PE [Facchinetti et al. 1999], although Staff and colleagues did not observe an antihypertensive effect when giving 12 g L-arginine orally for 2 days [Staff et al. 2004].
Given our observation that sFlt-1 is a direct determinant of ET-1 expression [Verdonk et al. 2015], sFlt-1 removal might be beneficial as well. Thadhani and colleagues accomplished this by apheresis in three women with severe early-onset PE, making use of a negatively charged dextran sulfate cellulose column capable of adsorbing sFlt-1. In their study, apheresis not only decreased circulating sFlt-1, but also normalized proteinuria, stabilized blood pressure and prolonged pregnancy, without evident adverse outcomes in either mother or child [Thadhani et al. 2011]. To what degree it affected ET-1 was not investigated. Given these promising findings, the results of other ongoing (interventional) studies applying the same approach are anxiously awaited.
As discussed above, ET receptor antagonists displayed beneficial effects in both PE animal models and rats treated with sunitinib. Currently, macitentan and bosentan, oral agents with dual (ETA and ETB) receptor blockade function, and the ETA receptor-selective antagonist ambrisentan, are available for clinical use. Beneficial effects of such drugs have been shown in pulmonary arterial hypertension, cancer and renal failure [Humbert et al. 2004]. At this stage, we do not know whether we should block both ET receptors in PE or only one subtype. Since ETB receptors also serve as clearance receptors [Kohan, 1997], selective ETB receptor antagonists, like ETB receptor knockout approaches [Gariepy et al. 2000], will increase circulating ET-1, thereby potentially elevating blood pressure. Selective ETA receptor antagonists therefore seem the preferred type of blocker, and additional ETB receptor blockade may or may not have additional beneficial effects. Future studies comparing dual and selective ETA receptor blockers head to head in appropriate models should answer this question.
Unfortunately, even when future animal studies will have yielded the best approach to block ET-1 (dual or single blockade), antagonism of ET receptors in pregnant women with PE may not be feasible, because of the potential teratogenic effects of such drugs [Clouthier et al. 1998; Treinen et al. 1999; Taniguchi and Muramatsu, 2003]. Fetal malformations have been observed in both ETA receptor knockout mice and rats treated with ETA receptor antagonists [Kurihara et al. 1994; Clouthier et al. 1998; Yanagisawa et al. 1998b]. Yanagisawa and colleagues observed that ETA receptor antagonists given to rodents early in pregnancy resulted in craniofacial anomalies and fetal death [Yanagisawa et al. 1998b]. This appeared not to be the case when administration occurred late in pregnancy [Thaete et al. 2001; Olgun et al. 2008; Reichetzeder et al. 2014]. In humans, Bédard and colleagues have also shown that pregnant women diagnosed with pulmonary arterial hypertension treated with ET receptor antagonist developed adverse effects, like premature delivery and neonatal mortality [Bédard et al. 2009]. Clearly therefore, if pursuing this pathway, we need drugs that do not cross the placental barrier, or novel approaches that selectively annihilate ET-1 in the mother, by suppressing endothelial ET-1 synthesis (e.g., with small interfering RNA), by blocking endothelial ET-1 release, or by binding or inactivation of ET-1 in the circulation.
Conclusions
Advances in our understanding of the pathophysiology of PE confirm PE as a complex multifactorial disease potentially requiring therapeutic intervention at multiple levels. The ET system now emerges as a final pathway that may be the cause of the hypertension, renal toxicity and RAS suppression in PE. Its blockade may therefore be beneficial, although simultaneously we know that ET receptor antagonism is teratogenic. Thus, such treatment may only be feasible if started at a stage sufficiently late to no longer allow teratogenic effects, or by making use of approaches that do not affect the fetus. We also need to know which ET receptor (A or B, or both) needs to be blocked. Alternatively, one might focus on the cause(s) of the ET-1 elevation (e.g. sFlt-1). This may yield new treatment tools, for instance sFlt-1 removal by apheresis.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors declare that there is no conflict of interest.