
Abstract
Objective:
The objective of this study was to determine the molecular mechanisms by which cardiac Rho-associated coiled-coil containing protein kinase (ROCK) activation after myocardial infarction (MI) does intervene in cardiac systolic function decline and remodeling.
Methods:
Simultaneous measurement of different cardiac ROCK target proteins levels, in vivo left ventricular (LV) systolic function, myocardial fibrosis and hypertrophy in rats with MI under ROCK inhibition with fasudil.
Results:
Seven days after MI, the ventricular mass increased significantly by 5.6% in the MI group and was reduced with fasudil. LV systolic dysfunction improved significantly with fasudil whereas cardiac ROCK activation was reduced to sham levels. The ROCK inhibitor also reduced increased cardiac levels of both ROCK1 and ROCK2 isoforms, cardiomyocyte ROCK2 fluorescence levels and β-myosin heavy chain (MHC) levels in addition to myocardial collagen volume fraction decline. Compared with sham rats, troponin phosphorylation levels after MI were similar and ROCK inhibition reduced them. MI significantly increased phosphorylation levels of extracellular-signal-regulated kinase (ERK) 42 and ERK 44 by twofold and 63%, respectively, whereas in the fasudil-treated MI group these levels were similar to those in the sham group. MI significantly increased phosphorylated levels of the transcription factor GATA-4 and the ROCK inhibitor normalized them.
Conclusions:
LV systolic dysfunction after MI was strongly associated with cardiac ROCK activation and subsequent phosphorylation of ROCK target proteins that promote ventricular remodeling such as β-MHC and the ERK/GATA-4 pathway. ROCK inhibition with fasudil significantly improved systolic function, diminished myocardial fibrosis and normalized β-MHC and ERK/GATA-4 phosphorylation levels.
Introduction
Myocardial infarction (MI) is a main cause of heart failure (HF) and death in most countries. Cardiac modifications due to MI and subsequent remodeling are compensatory mechanisms to maintain tissue integrity and preserve cardiac function. When this early adaptive response is progressive, cardiac remodeling becomes adverse and induces structural modifications leading to ventricular dysfunction and HF [Parajuli et al. 2012; Sutton and Sharpe, 2000; Haudek et al. 2009]. In this setting, worsening of contractility is associated with direct modifications in cardiac contractile proteins, by post-translational modifications or by changes in the expression of different isoforms [Hamdani et al. 2008].
During the acute phase after MI, the release of inflammatory mediators activates several intracellular signaling pathways. These pathways include neurohormonal responses that restore blood pressure and flow, alter the expression of myocardial proteins or modify the phosphorylation level of contractile proteins and in this way their function [Walker et al. 2010]. One of the signal transduction pathways associated with pathological remodeling and contractile dysfunction after MI is the RhoA signaling pathway and its main target protein, Rho-associated coiled-coil containing protein kinase (ROCK). The small GTPase Rho is activated by agonists of receptors coupled to the cell membrane G protein such as angiotensin II (Ang II) and noradrenaline. Activated Rho translocates to the cell membrane, where it turns on ROCK. Activated ROCK mediates several cellular functions such as muscle contraction, actin cytoskeleton organization, adhesion and motility, proliferation, differentiation, apoptosis, and the survival and expression of genes involved in cardiovascular remodeling [Jalil et al. 2005; Surma et al. 2011; Satoh et al. 2011; Loirand et al. 2013; Shi and Wei, 2013; Shi et al. 2011]. ROCK also mediates upregulation of several proinflammatory, thrombogenic and fibrogenic molecules, and downregulates endothelial nitric oxide synthase. In addition, ROCK activation promotes vascular oxidative stress [Rivera et al. 2007].
Experimental studies suggest that ROCK activation is involved in key processes associated with remodeling after myocardial injury and ventricular function decline [Hattori et al. 2004; Li et al. 2012a; Wang et al. 2011; Vargas et al. 2009; Rikitake et al. 2005; Shi et al. 2010; Jiang et al. 2013; Demiryürek et al. 2005; Zhang et al. 2010]. In mice overexpressing Gαq, ROCK1 gene deletion prevents ventricular dilatation and systolic dysfunction [Shi et al. 2008]. Furthermore, in cultured cardiomyocytes, RhoA/ROCK activation upregulates Bax through p53 to induce mitochondrial death pathway and cardiomyocyte apoptosis [Del Re et al. 2007]. In transgenic mice overexpressing the ROCK target myosin phosphatase target subunit-2, a higher phosphatase activity causes left ventricular (LV) dysfunction and enlargement [Mizutani et al. 2010].
Modifications in the regulatory proteins of cardiac contractility are associated with remodeling and impaired function in several heart diseases [Hamdani et al. 2008; Vahebi et al. 2005]. However, it is not known whether ROCK activation after MI is able to induce post-translational changes in the regulatory protein complex of contractility or whether its activation does regulate the expression of protein isoforms related to cell contraction and remodeling.
The aim of this study was to assess relevant molecular mechanisms by which cardiac ROCK activation after MI does intervene in cardiac systolic function decline and remodeling by measuring simultaneously the levels of cardiac ROCK target proteins, in vivo LV systolic function as well as myocardial fibrosis and hypertrophy in rats under pharmacological ROCK inhibition.
Methods
This work was performed according to the ‘Guide for the Care and Use of Laboratory Animals’ published by the National Health Institute (NIH No. 85-23, revised 1996) and was reviewed by the Ethics Committee on Animal Welfare of the Faculty of Medicine of Pontificia Universidad Católica de Chile. All efforts were made to reduce the number of animals used and their suffering.
Experimental MI model
Adult male Sprague-Dawley rats (weight 200 ± 10 g, aged 9 weeks) were obtained from our central facility. Rats were kept under a 12-hour light/dark cycle in a facility under both temperature and humidity regulated. Animals had free access to food and water ad libitum.
MI was induced by ligation of the left anterior descending artery (by left thoracotomy) under intraperitoneal (ip) anesthesia with ketamine (50 mg/kg) and xylazine (10 mg/kg) and mechanical ventilation. In the sham group, surgery was performed in the same way but without coronary artery ligation. MI was confirmed by echocardiogram 24–48 hours after surgery and infarcted rats were randomly divided into 2 groups: with or without oral treatment with fasudil, the most commonly used pharmacologic ROCK inhibitor (100 mg/kg/day by gavage for 1 week). Treatment with fasudil was started 1 day after surgery.
Two-dimensional (2D) echocardiographic parameters of LV systolic function
A week after coronary ligation, an echocardiographic study was blindly performed by an experienced echocardiographist under ketamine (35 mg/kg ip) and xylazine (7 mg/kg ip) with a Sonos 5500 instrument (Philips, Netherland) and a 12 MHz transducer (S12). A parasternal short axis view from the left ventricle was obtained and the following LV parameters were determined: end systolic diameter (ESD), end diastolic diameter (EDD), anterior wall thickness (AWT), posterior wall thickness (PWT), shortening LV fraction (SF), endocardial end systolic area (ESA) and endocardial end diastolic area (EDA). Fractional area change and ejection fraction (EF) were estimated based on these data according to conventional methodology [de Simone et al. 1996].
Morphometrical analysis
Myocardial hypertrophy and fibrosis were determined by morphometry in the left ventricle. One week after MI, rats were sacrificed by deep anesthesia. The hearts were washed in saline, weighted and fixed in 4% formalin in phosphate-buffered saline (PBS) for 12 hours and embedded in paraffin. Fixed hearts were transversally cut in 5 µm sections and stained with hematoxylin–eosin (HE) for morphological analysis of hypertrophy and with Picrosirius red for fibrosis analysis.
Cardiomyocytes cross-sections were examined in the noninfarcted LV wall under light microscopy (Nikon Eclipse E400) at 400× magnification. A total of 100 randomly selected cells were measured from each animal throughout the LV myocardium and endocardium. From each selected cell, the perimeter was delimited to calculate the cross-sectional area using the software NIS-Element BR 3.0.
Images for measuring interstitial collagen volume fraction (CVF) were acquired on an optical microscope (Nikon Eclipse E400) and were processed digitally to calculate CVF. For each LV, 20 pictures corresponding to the infarcted area, the area close to the infarct (defined as the area immediately adjacent to the scar) and the area far from the infarct (defined as the zone in the cross-section located exactly 180º from the middle of the infarct scar) were examined at 200× magnification. Collagen was quantified by computer-assisted morphometry using MATLAB 6.1 software.
Western blot analysis
For western blot analysis, samples from the entire LV were frozen in liquid nitrogen and stored at - 80°C until processing. Proteins were extracted from fresh-frozen myocardium. The entire LV samples were homogenized and lysed with lysis buffer with low concentrations of detergent [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) 50 mM, NaCl 150 mM, MgCl2 2 mM, ethylene glycol tetraacetic acid (EGTA) 1mM, Triton 1% and glycerol 10%) supplemented with protease inhibitors [aprotinin 2 µg/ml, leupeptin 10 µg/ml, phenylmethylsulfonyl fluoride (PMSF) 1mM] and phosphatases inhibitors (NaP2O7 4.46 mg/ml, NaF 10 mM, Na3VO4 1mM) on ice.
Equal amounts of protein (25 µg) were loaded and separated on a 10% sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE) gel and transferred to a nitrocellulose membrane (Bio-Rad). After blocking with 7% nonfat milk (for nonphosphorylated proteins) or bovine serum albumin (BSA) 5% (for phosphorylated proteins) for 1 hour at room temperature, the blots were incubated overnight at 4°C with the following antibodies: anti-myosin phosphatase target subunit-1 (MYPT-1, mouse monoclonal, 1/1000 BD Biosciences, Cat 612164); p-MYPT-1 (phospho-MYPT1-Thr 853 rabbit polyclonal, 1/700, Cyclex Co, Cat CY-P1025), ezrin–radixin–moesin (ERM, total-ERM, rabbit polyclonal, 1/700, Cell Signaling, Cat 3142S); p-ERM (phospho-ERM, Ezrin Thr567, Radixin Thr564, Moesin Thr558, rabbit polyclonal, 1/700, Cell Signaling, Cat 3141F); β-MHC (myosin heavy chain, mouse monoclonal, 1/1000, Novocastra TM); α-SKA (α-skeletal actin, mouse monoclonal, 1/2500, US Biological Life Sciences, Cat A0760-24); p-SMAD3 (phospho-SMAD S423/425, rabbit monoclonal, 1/1000, Cell Signaling, Cat 8769S); SMAD3 (total-SMAD, rabbit monoclonal, 1/1000, Cell Signaling, Cat 9523S); ROCK-1 (mouse monoclonal, 1/1000, BD BioScience, Cat 611136); ROCK-2 (mouse monoclonal, 1/1000, BD BioScience, Cat 610623); troponin I (cTnI, total-troponin I, mouse monoclonal, 1/1000, Abcam, Cat Ab19615); p-cTnI (phospho-troponin I, phospho S22+S23, 1/1000, Rabbit polyclonal, Abcam, Cat Ab58545); ERK1/2 (total extracellular-signal-regulated kinase, rabbit polyclonal, 1/1000, Cell Signaling, Cat 9102); p-ERK1/2 (phospho-ERK 44/42 Thr202/Tyr204, 1/1000, Cell Signaling, Cat 9101); GATA-4 (total-GATA-4, rabbit polyclonal, 1/1000, Thermo Scientific, Cat PA592663); and p-GATA-4 (Phospho-GATA-4, phospho S105, 1/1000, rabbit polyclonal, Abcam, Cat 5245). The blots were then washed and incubated with a secondary antibody horseradish peroxidase (HRP) conjugated goat anti rabbit immunoglobulin (Ig) G (1:5000, Thermo Scientific) or a goat anti mouse IgG (1:10.000, Santa Cruz) for 2 hours.
The relative amount of protein was estimated by chemiluminescence using the ECL plus kit (Perkin Elmer), which contains the substrate for HRP. Digital images obtained from the photographic films were analyzed by densitometry using the software Image J (NIH, USA). A glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mouse monoclonal antibody (1:1000) (Santa Cruz Biotechnology Inc.) was used as a protein loading control.
ROCK2 immunofluorescence
For immunofluorescence, 5 µm sections were transversally cut from fixed hearts. Immunolabeling was performed using an anti-ROCK2 antibody (mouse, 1:400, Abcam). Alexa Fluor® 488 anti-mouse IgG (1:1000) was used as the secondary antibody. These sections were also stained with wheat germ agglutinin (WGA) conjugated with Alexa 594 (Molecular Probes) to define and evaluate the integrity of the plasma membrane of cardiomyocytes. WGA is a carbohydrate-binding protein that selectively recognizes sialic acid and N-acetylglucosaminyl sugar residues found in the plasma membranes. Images were obtained using a Fluoview FV1000 confocal microscope (Olympus 1x2-UCB) with a 60× objective. Each section of the zone of interest in the LV underwent a sweep along the z axis every 0.5 µm, yielding a total of approximately 15–20 images (stack). In order to quantify the fluorescence signal, a reconstruction was performed on the z axis to determine the average intensity of the interest signal (average intensity). To obtain high-contrast images a reconstruction was made on the z-axis signals with maximum intensity (maximal intensity). This analysis was made using the software Image J (NIH, USA).
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM). Differences between the mean values were compared by one-way analysis of variance (ANOVA) followed by the Newman–Keuls test. A p value < 0.05 was considered statistically significant.
Results
Characteristics of the experimental groups and LV function data (Table 1)
One week after MI, body weight was reduced in both the MI group and the MI + fasudil group compared with the sham operated animals, but it was not statistically significant and might suggest a degree of cachexia with wasting of soft tissues. Ventricular mass (VM), corresponding to the weight of both ventricles, was significantly increased in rats with MI compared with the sham group by 6%, whereas in fasudil-treated MI rats, VM was lessened significantly by 18% compared with the untreated infarcted rats (and by 13% compared with the sham group). The relative ventricular mass (RVM) (ventricular mass/body weight) increased significantly by 30% in the MI group compared with the sham group and it was attenuated by 17% with fasudil.
Characteristics of the experimental groups.

Data are presented as mean ± standard error of the mean.
p < 0.05 versus sham; $p < 0.05 versus MI (Newman–Keuls, after significant one-way analysis of variance)
AWT, anterior wall thickness; EDA, end diastolic area; EDD, end diastolic diameter; EF, ejection fraction; ESA, end systolic area; ESD, end systolic diameter; IA, infarcted area; MI, myocardial infarction; RVM, relative ventricular mass; SF, shortening fraction; VM, ventricular mass.
LV dimensions and systolic function assessed by echocardiography (Table 1)
In the MI group, an important dilatation of the LV cavity was observed. End systolic LV area was significantly increased by 2.6 times in the MI group compared with the sham group and it was diminished by 40% with fasudil. At the same time, the shortening fraction (SF) and ejection fraction (EF) were both severely compromised in the untreated infarcted rats. Worsening in the LV systolic function was significantly improved by pharmacological ROCK inhibition for 7 days.
Activation of the RhoA/ROCK signaling pathway
Cardiac ROCK activation was assessed by determining phosphorylation levels of its two major substrate proteins, MYPT-1 and ERM in the noninfarcted LV myocardium.
Phosphorylation of MYPT-1 (p-MYPT1) in amino acid residue 853 is specific for ROCK [Ito et al. 2004]. In rats with MI, p-MPYT1 was significantly increased by 2.9 ± 0.3 fold compared with the sham group and it decreased significantly with fasudil administration

In order to better precise cardiac ROCK activation 1 week after MI, phosphorylation levels of a second ROCK target, ERM, were determined in the entire LV myocardium. In the infarcted rats a significant increase of cardiac p-ERM levels by 2.2 ± 0.14 fold compared with the sham group was observed. ERM levels were significantly reduced in the MI + fasudil group (Figure 2).

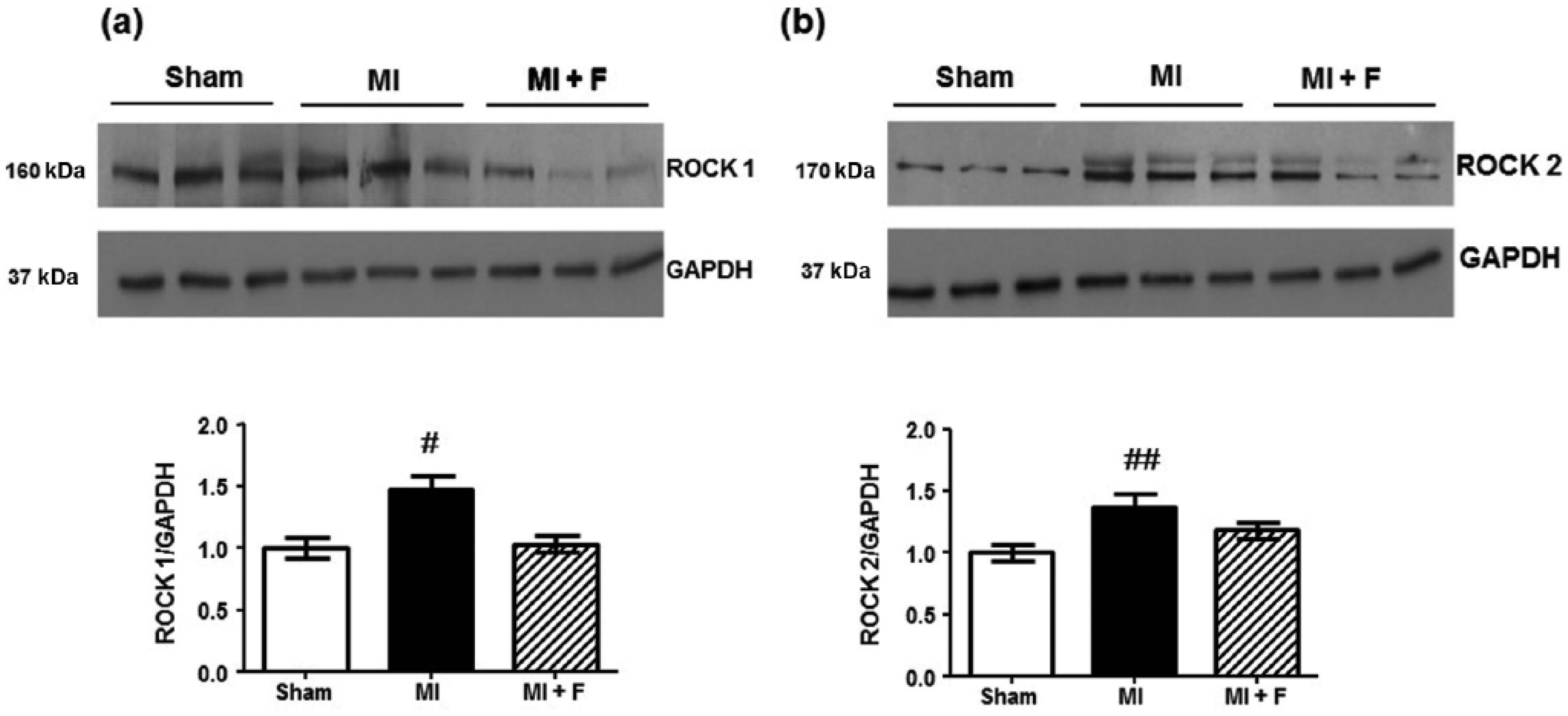
Levels of cardiac ROCK isoforms
Protein levels of ROCK1 and ROCK2 isoforms in cardiac tissue after MI were measured by western blotting. A significant increase in the levels of both isoforms was observed (Figure 3a and Figure 3b). Myocardial levels of ROCK1 increased by 50% (p < 0.01) and levels of ROCK2 increased by 40% (p < 0.05) 1 week after MI. Fasudil significantly reduced ROCK1 levels similar to the control group. ROCK2 cardiac levels were also reduced with fasudil to 20% over control animals.
ROCK2 immunolocalization
Protein levels of each ROCK isoform were determined in the entire LV homogenate, which contains several cell types (cardiomyocytes, fibroblasts and endothelial cells). In order to examine whether these changes were observed mainly in cardiomyocytes, immunofluorescence analysis for ROCK2 was performed on transversally cut sections from fixed hearts obtained 1 week after MI.
Cardiomyocyte membranes were visualized by WGA staining, demonstrating the integrity and preservation of the cardiomyocyte cell membrane.
Intensity fluorescence levels for ROCK2 were significantly increased in the infarcted rat cardiomyocytes by 2.6 fold compared with the control group. Fasudil significantly reduced ROCK2 intensity fluorescence to levels observed in the control group (Figure 4).

Cardiomyocyte cross-sectional area, β-MHC and α-SKA levels
Cardiomyocyte hypertrophy (CH) was assessed by the levels of the protein markers β-MHC and α-SKA in the entire LV (by Western blot) and by morphometry.
Protein levels of β-MHC and α-SKA were used as early markers of LV hypertrophy. A period of 7 days after MI, β-MHC myocardial levels were increased by 30 fold compared with the sham group (Figure 5b) whereas fasudil significantly reduced β-MHC myocardial levels to 3.4 fold over the sham group (p < 0.001). Similarly, in rats with MI, protein levels of α-SKA were increased by 85% (p < 0.001) compared with the sham group (Figure 5c) without changes after fasudil administration.

Mean cardiomyocyte cross-sectional area in the sham group was 167 ± 3 µm2; it was significantly larger by 10% in the MI group (p < 0.05). Fasudil administered to the MI group for 7 days did not change the mean cardiomyocyte cross-sectional area (p < 0.05 compared with the sham group).
Myocardial fibrosis and SMAD3 levels
Myocardial fibrosis was assessed by measuring interstitial collagen morphometrically. Both in the areas close to MI and in the areas far from MI a significant increase in CVF was observed in the MI group compared with the sham-operated group (1.16 ± 0.34 versus 5.1 ± 0,6 % and 1.16 ± 0.34 versus 4.2 ± 0.3 %; n = 6, p< 0.001, respectively) (Figure 6). Myocardial fibrosis was attenuated in the fasudil-treated MI group in both LV areas compared with the untreated MI group.

Phosphorylation levels of SMAD3, a downstream target in the activation of the TGF-β signaling pathway involved in fibrosis, was measured. SMAD3 phosphorylated levels were similar both in the MI and in the sham groups, but a significant increase in total SMAD3 protein levels was observed in the MI compared with the sham group, by 65% (Figure 7). Fasudil significantly reduced total SMAD3 protein levels to those observed in the sham group.

cTnI phosphorylation levels
Due to its role in cardiac contractile function and possible modifications induced by cardiac ROCK activation, cTnI levels were determined here by western blot. Compared with sham rats, MI cTnI phosphorylation levels were similar (Figure 8). Interestingly, in the fasudil-treated MI group both phosphorylated and total cTnI levels were significantly reduced by 75%.

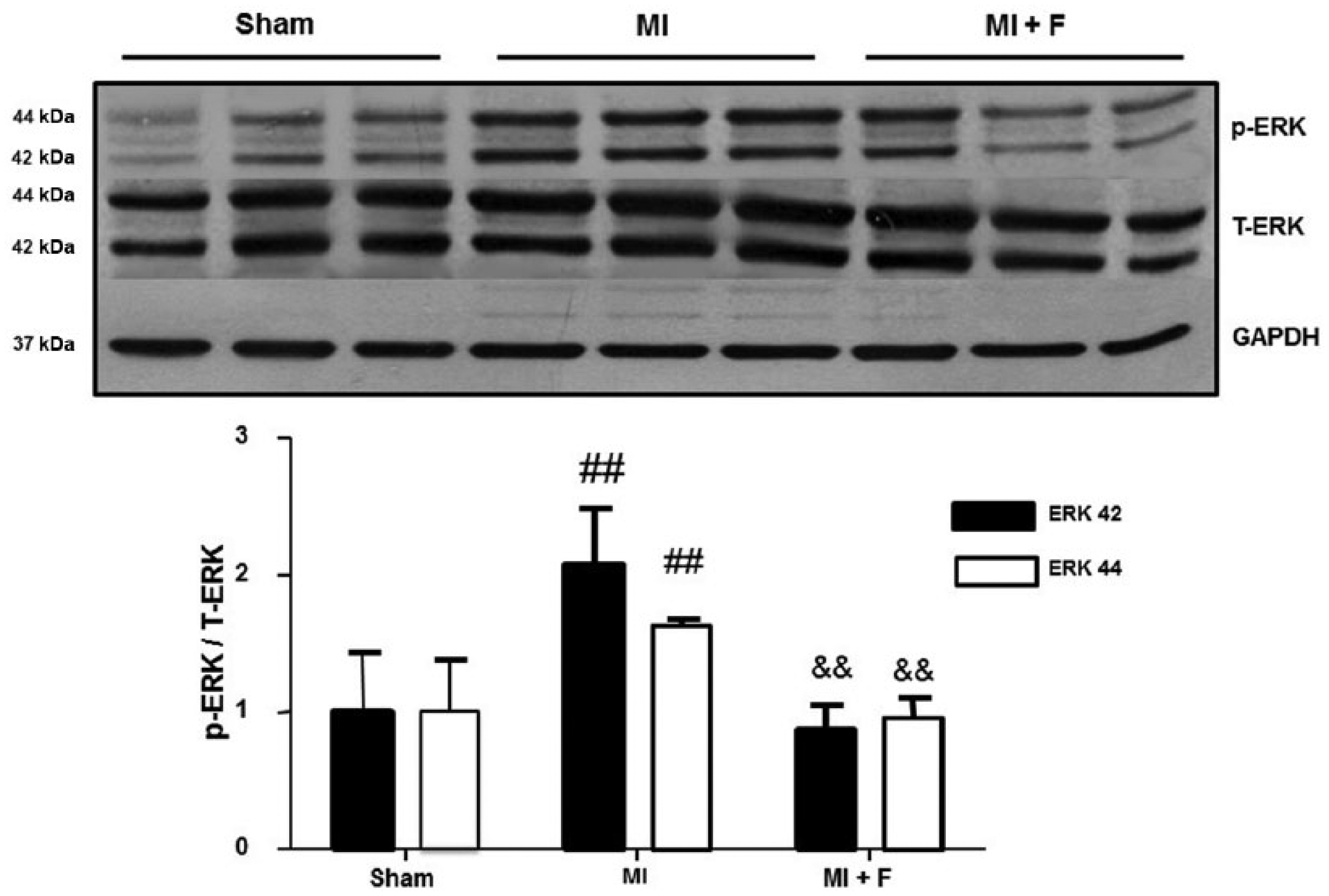
ERK 42, ERK 44 and GATA-4 cardiac phosphorylation levels
Because of the role played by the signaling pathway ROCK/ERK/GATA-4 on myocardial remodeling and contractility, their levels were measured. MI significantly increased phosphorylation levels of ERK 42 and ERK 44 by 2 fold and 63%, respectively (Figure 9), whereas in the fasudil-treated MI group these levels were similar to those observed in the sham group.
In addition, MI significantly increased phosphorylated levels of the transcription factor GATA-4 by 2.9 fold (Figure 10), whereas fasudil reduced them to levels similar to those in the sham group. MI also induced a significant increase in total protein levels of GATA-4 by 2.5 fold without changes in the fasudil-treated MI group.

Discussion
The main observations in this study, in a 1 week MI model in the rat, were that the development of LV systolic dysfunction was strongly associated with cardiac ROCK activation and phosphorylation of ROCK target proteins that promote ventricular remodeling and LV systolic dysfunction such as β-MHC and the ERK/GATA-4 pathway. ROCK inhibition with fasudil significantly improved systolic dysfunction and diminished myocardial fibrosis, which is consistent with other studies [Chau et al. 2011, Hattori et al. 2004]. Besides, ROCK inhibition normalized β-MHC and ERK/GATA-4 phosphorylation levels. The detected increase of both ROCK cardiac isoforms and increased ROCK2 immunolocalization within the cardiomyocyte after MI (as well as reduction with fasudil) are the first observations in systolic dysfunction due to MI.
ROCK activation and systolic dysfunction after MI
Currently, the most well recognized method to determine ROCK activation is by measuring the phosphorylation level of one of its downstream target proteins, MYPT1. MYPT1 is part of the holoenzyme complex myosin phosphatase, the regulatory subunit of myosin binding. This phosphatase activity is regulated by the phosphorylation state of threonine residues 696 and 853 in MYPT1, which can be phosphorylated by ROCK [Ichikawa et al. 1996; Feng et al. 1999; Khromov et al. 2009]. In vitro studies have established that phosphorylation at residue 853 is completely prevented by ROCK inhibitors, while the threonine 696 residue can also be phosphorylated by other kinases.
Here, in rats with early systolic dysfunction 1 week after MI, cardiac MYPT1 phosphorylation levels were significantly increased and the ROCK inhibitor fasudil reduced MYPT1 phosphorylation, indicating the role of cardiac ROCK activation after MI. In order to better characterize ROCK phosphorylation specificity in this model, the cardiac ERM phosphorylation level, another ROCK target [Shi et al. 2011; Hébert et al. 2008], was determined in the noninfarcted LV myocardium in a sample from the three experimental groups as observed in mice [Chau et al. 2011]. A significant increase in cardiac ERM phosphorylation levels was observed 1 week after MI and it was also decreased by fasudil. Our ERM findings are consistent with observations in mice on cardiac ROCK activation 4 days [Chau et al. 2011] and 4 weeks after MI [Hattori et al. 2004], as well as in the ischemia reperfusion model in the rat [Li et al. 2012b]. ERM proteins are closely related proteins originally identified as cytoskeleton crosslinkers and important components of cell structure [Niggli and Rossy, 2008; Darmellah et al. 2009], but the role of ERM phosphorylation in cardiac cells is not well known. In cardiomyocytes, activated ERM proteins mediate the effects of acid-induced Na+/H+ exchanger (NH) activation and Akt is a downstream effector in the cascade activated by NH–ERM interaction [Darmellah et al. 2009].
In addition, a significant increase in both cardiac isoforms ROCK1 and ROCK2 was observed here for the first time in this experimental model. Increased ROCK1 and ROCK2 isoforms have been detected experimentally in vascular smooth muscle cells in experimental hypertension and also in pulmonary arteries in hypoxia and after lung damage [McNamara et al. 2008]. A simultaneous increase in both cardiac ROCK isoforms was observed in rats with streptozotocin-induced diabetes [Guo et al. 2013b] and also after 24 hours in the isquemia-reperfusion model [Guo et al. 2013]. Besides, we localized increased ROCK2 levels (and the effect of the ROCK inhibitor) within the cardiomyocytes using immunofluorescence for the first time in this MI model, in parallel with β-MHC augment and supporting the role of ROCK2 in cardiac hypertrophy.
Ventricular remodeling
ROCK1 contributes to the pathologic remodeling events including cardiac fibrosis, cardiomyocyte apoptosis and contractile dysfunction, leading to cardiac dilation and HF, and it appears to mediate impaired ERK- and Akt-dependent survival signaling and increased fibrogenic cytokines and altered adenylyl cyclase (AC) 5/6 expression induced by hypertrophic stimuli [Shi et al. 2011]. However, genetic studies using cardiac-specific ROCK2 knockout mice have demonstrated that ROCK2 is involved in cardiomyocyte hypertrophy, apoptosis and cardiac fibrosis during compensatory cardiac hypertrophy [Okamoto et al. 2013; Shi and Wei, 2013].
Recent genetic studies indicate that ROCK isoforms may have distinct roles in cardiac remodeling. But whether the beneficial effects of ROCK inhibitors are mediated by inhibition of ROCK1, ROCK2 or both remains to be determined. Further study of conditional knockout mice of ROCK1 and ROCK2 as well as the use of isoform-specific inhibitors would provide novel insights into their roles in MI and remodeling.
Myocardial injury is associated with re-expression of genes normally expressed early during fetal life such as β-MHC and α-SKA, which are early hypertrophy markers [Sutton and Sharpe, 2000; Yanazume et al. 2002; Akazawa and Komuro, 2003]. In our rats with MI, a significant increase in cardiac levels of β-MHC and α-SKA was observed, consistent with previous reports [Sutton and Sharpe, 2000; Yanazume et al. 2002; Akazawa and Komuro, 2003]. Fasudil reduced β-MHC increase to normal levels, indicating that ROCK activation contributes to remodeling additionally through β-MHC expression. Fasudil failed to reduce cardiac α-SKA levels, suggesting that the expression of this protein after MI is independent of ROCK activation.
In cardiac hypertrophy due to pressure overload, two distinct myocyte populations may exist within the same hypertrophic heart: one that is hypertrophic and predominantly expresses α-MHC and a second that is not hypertrophic and expresses both α- and β-MHC [Lopez et al. 2011; Pandya and Smithies, 2011]. It has been proposed that β-MHC re-expression is a marker of cellular ‘normotrophy’ under conditions that induce overall cardiac hypertrophy and the increase in total MHC that results in the β-MHC myocytes makes them resistant to increases in cell size so that the induction of β-MHC is in fact protective [Lopez et al. 2011; Pandya and Smithies, 2011]. Additional findings show that re-expression of β-MHC occurs in discrete subsets of myocytes within the subendocardium and suggest that β-MHC induction is not an obligatory consequence of cellular hypertrophy and that cells expressing β-MHC in the hypertrophic heart are distributed predominantly in clusters within and surrounding foci of fibrosis [Pandya et al. 2006].
To further explore the mechanism by which ROCK activation does regulate the expression of cardiac β-MHC, we focused on the transcription factor GATA-4, which is able to translocate to the nucleus and binding to consensus sequences [(A/T)GATA(A/G)] in the gene promoters encoding proteins such as β-MHC, angiotensin II type 1 receptor, endothelin-1 [Guo et al. 2013] and also cTnI [Murphy et al. 1997]. Transcriptional activation of GATA-4 is dependent on phosphorylation on serine residue 105 and is mainly mediated by mitogen-activated protein kinase kinase (MEK) 1 [Cai et al. 2009] and ERK 1/2 [Wang et al. 2011; Yanazume et al. 2002; Akazawa and Komuro, 2003]. ROCK is a direct regulator of the ERK1/2 pathway, which does phosphorylate and activate cardiac GATA-4 [Yanazume et al. 2002]. We observed an increase in ERK1/2 phosphorylation levels in MI rats, as well as enhanced phosphorylation at serine residue 105 in cardiac GATA-4. Both changes were reduced with fasudil, indicating the role of ROCK in these processes. Activation of the cardiac ROCK/ERK/GATA-4 pathway here was also consistent with the increase in the expression of β-MHC in the MI group.
Along with cardiac hypertrophy, in this model of severe MI, increased interstitial collagen deposition was observed in the noninfarcted LV. Fasudil reversed myocardial fibrosis, demonstrating the key role of ROCK activation in myocardial fibrosis [Hattori et al. 2014; Li et al. 2012a; Wang et al. 2011; Vargas et al. 2009; Rikitake et al. 2005; Chau et al. 2011]. In mice with ROCK1 gene deletion, the role of this isoform has been identified in the development of cardiac fibrosis in response to isquemia reperfusion [Haudek et al. 2009] and in ROCK1(+/-) haploinsufficient mice stimulated with angiotensin II [Rikitake et al. 2005].
It has been proposed that ROCK can regulate the release of pro-inflammatory cytokines involved in fibrosis such as interferon-γ, monocyte chemotactic protein 1 (MCP 1) and transforming growth factor (TGF) β [Hattori et al. 2004]. TGFβ is expressed in the heart, and it has been shown that both cardiomyocytes and fibroblasts release TGFβ1 [Hao et al. 1999]. The canonical pathway of TGFβ activation involves activation of intracellular SMAD proteins, which are transcription factors regulating gene expression of extracellular matrix proteins [Bujak et al. 2007; Kamato et al. 2013]. Thus, we examined cardiac SMAD3 phosphorylation levels as a participant in the TGFβ activation pathway [Kamato et al. 2013; Zavadzkas et al. 2011], but its levels were similar to controls.
Cardiac ROCK and regulation of contractile proteins after MI
Treatment with fasudil for 1 week after MI induced a significant improvement in cardiac hypertrophy and fibrosis as well as in LV systolic function. To understand whether ROCK is able to interact with proteins of the cardiac contractile apparatus, cTnI phosphorylation levels were determined. Despite cardiac ROCK activation, phosphorylation levels of cTnI in ser23/ser24 were similar to those in control animals. In mice with MI, cardiac (papillary muscle) troponin I phosphorylation and myosin light chain 2 are reduced after MI [Avner et al. 2012]. Besides, in our MI rats treated with fasudil, a reduction in cTnI total protein levels was observed which might suggest that ROCK inhibition after MI does regulate expression of this contractile protein. It is not possible to rule out as an alternative explanation for these findings, that is, an increased cTnI turnover induced by ROCK inhibition after MI.
The current in vivo results, increased cardiac phosphorylation of ROCK target proteins and its reduction with fasudil, along with LV systolic function improvement demonstrate that the observed effects are specific for this kinase. Besides, the increase in cardiac protein levels of MYPT1, β-MHC and SMAD3 and the subsequent decline to those in sham levels with fasudil, suggests that this kinase is an important regulator in these post-translational modifications of proteins as well as in transcription associated with remodeling and contractility [Berenjeno and Bustelo, 2008; Peng et al. 2012; Vlasblom et al. 2009].
Limitations
Despite the usefulness of the in vivo model and since the induced cardiac changes were observed in an integrated pathophysiological context, the role of other signaling pathways was not assessed here. Furthermore, the current observations, 1 week after MI, might not be present in the long term. However, previous data in mice with MI at 4 weeks strongly suggest that the deleterious effects of ROCK activation on remodeling and LV function are more than transient [Hattori et al. 2004]. Besides, we used the entire LV rather than the noninfarct region of the LV for molecular biochemical determinations. However, for the purposes of interpretation of the results, the same measurements were performed in both MI experimental groups with similar infarct areas, but in the MI group more remodeling in the noninfarcted zone compared with the MI + fasudil treated group was observed. Thus, it is reasonable to propose that the observed molecular changes most probably took place in the noninfarcted area. In addition, no gene expression studies were performed, but the role of several important phosphorylated proteins participating in LV remodeling and function was analyzed.
In conclusion, relevant mechanisms by which early ROCK activation after MI does deteriorate LV systolic function are hypertrophy and fibrosis promotion, and phosphorylation of cardiac ERK 42, ERK 44 and transcription factor GATA-4. ROCK inhibition after MI significantly improves LV systolic function and simultaneously does normalize all these altered mechanisms.
Footnotes
Acknowledgements
We are grateful to Soledad Veliz for her technical and dedicated work.
Funding
This work was supported by Fondecyt (grant numbers 1085208 and 1121060 to J.J.), Fondap (grant number 15130011 to M.P.O.), a Comisión Nacional de Investigación Científica y Tecnológica (CONICYT) fellowship (to C.M.) and a Young Investigator Sochicar grant (to C.M.).
Conflict of interest statement
The authors declare no conflicts of interest in preparing this article.