
Abstract
In angiotensin (Ang)-II-dependent hypertension, collecting duct renin synthesis and secretion are stimulated despite suppression of juxtaglomerular (JG) renin. This effect is mediated by Ang II type 1 (AT1) receptor independent of blood pressure. Although the regulation of JG renin is known, the mechanisms by which renin is regulated in the collecting duct are not completely understood. The presence of renin activity in the collecting duct may provide a pathway for intratubular Ang II formation since angiotensinogen substrate and angiotensin converting enzyme are present in the distal nephron. The recently named new member of the renin–angiotensin system (RAS), the (pro)renin receptor [(P)RR], is able to bind renin and the inactive prorenin, thus enhancing renin activity and fully activating prorenin. We have demonstrated that renin and (P)RR are augmented in renal tissues from rats infused with Ang II and during sodium depletion, suggesting a physiological role in intrarenal RAS activation. Importantly, (P)RR activation also causes activation of intracellular pathways associated with increased cyclooxygenase 2 expression and induction of profibrotic genes. In addition, renin and (P)RR are upregulated by Ang II in collecting duct cells. Although the mechanisms involved in their regulation are still under study, they seem to be dependent on the intrarenal RAS activation. The complexities of the mechanisms of stimulation also depend on cyclooxygenase 2 and sodium depletion. Our data suggest that renin and (P)RR can interact to increase intratubular Ang II formation and the activation of profibrotic genes in renal collecting duct cells. Both pathways may have a critical role in the development of hypertension and renal disease.
Introduction
Although the renin–angiotensin system (RAS) plays a major role in the physiological control of blood pressure and fluid volume, it also potentially contributes to the development and progression of hypertension and fibrotic and hypertrophic diseases. Beside the systemic RAS, which is mainly controlled by the production and release of renin from the juxtaglomerular (JG) cells in the kidneys, local RAS present in a variety of organs is also important, particularly along the nephron. Renin is primarily synthesized in the JG cells where it is stimulated by a number of signals that increase intracellular cyclic adenyl mono-phosphate (cAMP) levels and activation of protein kinase A (PKA) [Castrop et al. 2010]. The 5’-flanking noncoding region of the renin gene [Borensztein et al. 1994; Castrop et al. 2010] plays a central role in regulating renin expression in all species. This region is considered the classic renin gene promoter. The presence of a functional cAMP response element (CRE) is a characteristic feature of the renin promoter in all species; cAMP binds to the two regulatory subunits of PKA to release two catalytic subunits from the inactive PKA tetramer complex. The free catalytic subunits (referred to as activated PKA) translocate to the nucleus and phosphorylate transcription factors of the cAMP response element binding protein/activating transcription factor CREB.
In contrast to the stimulatory effect on cAMP, angiotensin (Ang) II inhibits renin gene expression and secretion from JG cells by increasing the cytosolic calcium and activating protein kinase C (PKC) [Muller et al. 2002]. The presence of prorenin and renin has been demonstrated in renal collecting ducts [Seikaly et al. 1990; Prieto-Carrasquero et al. 2004, 2005, 2008, 2009; Prieto et al. 2013; Rohrwasser et al. 1999, 2003]. Furthermore, it has been reported that Ang II increases renin expression in the collecting duct cells in vitro and in vivo [Gonzalez et al. 2011b] which is opposite to what has been observed in JG cells [Muller et al. 2002]. These findings are of great relevance in the view that angiotensinogen (AGT) and angiotensin-converting enzyme (ACE) are also present along the nephron and are also upregulated by chronic Ang II infusions [Kobori et al. 2001; Gonzalez-Villalobos et al. 2010], indicating that augmented collecting duct renin may contribute to further intratubular Ang II formation in the distal nephron segments.
With the discovery and characterization of the new member of the RAS, the (pro)renin receptor [(P)RR], a new spectrum of possibilities and pathways with potential roles in the pathogenesis of hypertension and kidney disease have been proposed. The (P)RR is an ATPase H(+)-transporting lysosomal accessory protein (ATP6AP2), but also acts as a membrane receptor of renin and prorenin. The binding of (P)RR to renin and prorenin enhances renin activity and fully activates the biologically inactive prorenin [Nguyen and Contrepas, 2008; Nguyen and Muller, 2010], thus contributing to further Ang I formation inside the kidneys [Nguyen et al. 2002]. Additionally, binding of prorenin and renin to the membrane bound (P)RR triggers intracellular pathways which have been related to tissue damage [Nguyen et al. 1996; Nguyen and Danser, 2006]. In this review we will discuss the evidence demonstrating that the principal cells of the collecting duct synthesize and secrete renin/prorenin in response to the physiological activation of RAS with low sodium diet, as well as to Ang II treatment. We will also deliberate about the presence of (P)RR in the intercalated cells and its interaction with renin/prorenin in the collecting duct and their possible role in regulating intrarenal Ang II levels during intrarenal RAS activation.
Intrarenal RAS: evidence of the augmented expression of AGT and ACE during hypertension
Ang I and Ang II concentrations in the proximal tubule fluid are in the range of 5–10 pmol/ml [Navar and Harrison-Bernard, 2000; Navar et al. 2001, 2002], which are similar to renal interstitial fluid concentrations [Nishiyama et al. 2001] and remain elevated in infused hypertensive rats [Wang et al. 2003], suggesting actions on proximal reabsorption rate. The presence of Ang II type 1 (AT1) receptors on luminal membranes of proximal and distal nephron segments suggested that Ang II concentrations are able to activate AT1 receptors [Peti-Peterdi et al. 2002; Komlosi et al. 2003]. In mice, AT1a receptors are essential for normal blood pressure regulation and for mediating the hypertensive response to Ang II infusions [Crowley et al. 2006]. These studies indicate that distal nephron Ang II is formed locally in the tubules at concentrations that are sufficiently high to influence distal nephron transport function [Peti-Peterdi et al. 2002; Komlosi et al. 2003].
AGT mRNA is present in the proximal tubule cells [Ingelfinger et al. 1999; Kobori et al. 2001, 2002, 2003]. This observation generated interest about its function. A breakthrough was the publication of several reports describing the augmentation of mRNA and urinary excretion in a chronic Ang II infusion model [Kobori et al. 2002, 2003]. This effect is mediated via activation of AT1 receptors, since AT1 receptor blockers prevented AGT upregulation [Kobori et al. 2004]. Similarly, in vitro studies using proximal tubule cell cultures show that Ang II stimulates AGT mRNA and protein by a mechanism that involves the interactions with inflammatory factors including interleukin [Satou et al. 2009] and oxidative stress [Satou et al. 2008, 2012]. The role of AGT in hypertension has been established using genetic models that lead to overexpression of AGT. AGT may be used as a marker, reflecting the intratubular RAS status that is correlated with kidney Ang II levels in Ang II-dependent hypertensive rats [Kobori et al. 2002, 2003]. Recently, direct quantitative methods to measure urinary AGT using human/mouse/rat AGT enzyme-linked immunosorbent assay have been developed [Kobori et al. 2008]. Using this system, urinary excretion rates of AGT have been used as an index of intrarenal RAS status in patients with chronic kidney disease [Yamamoto et al. 2007], diabetes mellitus [Ogawa et al. 2009], and hypertension [Lantelme et al. 2005; Kobori et al. 2010]. Importantly, patients treated with RAS blockers showed reduced urinary AGT levels [Kobori et al. 2003].
ACE is another key enzyme of the RAS that is also present in the collecting duct [Vio and Jeanneret, 2003]. This enzyme allows the conversion of most of the Ang I into Ang II and there is evidence demonstrating that ACE is augmented in Ang II-dependent hypertension [Harrison-Bernard et al. 2002; Gonzalez-Villalobos et al. 2009, 2010] and other models of kidney injuries [Vio and Jeanneret, 2003]. These findings, along with the demonstration that proximal tubule cells express AGT and that ACE activity is also present in the collecting ducts [Redublo Quinto et al. 2008; Casarini et al. 1997], support an important physiological role of tubular renin.
Prorenin and renin synthesis in the collecting duct cells is augmented by Ang II in vitro and during physiological RAS activation
Besides its primary localization in the JG cells, renin mRNA and protein expression is also present in renal tubules [Rohrwasser et al. 1999, 2003; Prieto-Carrasquero et al. 2004, 2005, 2008]. Renin synthesis is augmented in the principal cells of connecting tubules and cortical and medullary collecting ducts of chronic Ang II-infused rats and mice, Cyp1a1Ren2 transgenic rats, and in both kidneys of two-kidney one clip Goldblatt hypertensive rats [Rohrwasser et al. 1999, 2003; Prieto-Carrasquero et al. 2004, 2005, 2008].
In contrast to the inhibitory effect that Ang II exerts on JG renin, Ang II stimulates renin in the principal collecting duct cells via a mechanism mediated by the AT1 receptor independent of changes in blood pressure [Prieto-Carrasquero et al. 2004, 2005, 2008]. Activation of AT1 receptors suppresses renin synthesis in JG cells via PKC and Ca2+ [Kurtz and Wagner, 1999]; however, we demonstrated that augmentation of renin synthesis in rat inner medullary collecting duct (IMCD) cells is mediated directly by AT1 receptors via a PKC pathway [Gonzalez et al. 2011b]. Figure 1(a) shows renin immunostaining in a typical scattered pattern in isolated IMCD cells (arrows). In IMCD cultured cells, renin immunostaining is augmented after Ang II treatment (10−7 M) during 6 h. As shown in Figure 1(b), prorenin–renin bands can be identified in cell lysates of IMCD cells. Treatment with Ang II increases prorenin (195 ± 25%, p < 0.05) and renin (200 ± 20%, p < 0.05) abundance compared with controls. Treatment with PKC inhibitor, calphostin C, prevented the stimulatory effect of Ang II on prorenin (88 ± 12%, p = 0.56) and renin (95 ± 15%, p = 0.23). The phorbol ester phorbol 12-myristate 13-acetate (PMA), a PKC activator, greatly increases prorenin (172 ± 13%, p < 0.05) and renin (189 ± 15%, p < 0.05) compared with controls.

(a) Immunofluorescence localization of prorenin/renin (green) in rat primary cultured IMCD cells. Nuclei were counterstained with DAPI (blue). IMCD cells treated with Ang II (10−7 mol/L) increased prorenin/renin immunofluorescence, which was prevented in the presence of angiotensin type II receptor blocker candesartan (Cand) and calphostin C (Cal C). (b) Renin/prorenin protein expression levels in IMCD cells were augmented in the presence of Ang II, but Cal C prevented this response. The effect of Ang II on prorenin/renin protein levels was similarly observed in response to phorbol 12-myristate 13-acetate (PMA), a protein kinase C activator. Band identity was confirmed by using recombinant human renin and human pro)renin as standards.
The augmented levels of renin in the collecting duct in Ang II-dependent hypertensive rats may explain why new Ang II is formed intratubularly in this animal model of hypertension [Shao et al. 2009, 2010]. Recently, our group also showed that during low salt conditions (14 days), a physiological status of RAS activation, renin was augmented in medullary tissues [Shao et al. 2013]. In a recent publication we also demonstrated that a short period of 7 days of sodium depletion can also induce the expression of renin mRNA and protein in medullary renal tissues [Gonzalez et al. 2014b], suggesting that renin synthesis is stimulated during a low-sodium diet. These observations suggest that the induction of intrarenal RAS may promote further Ang II acting directly to enhance sodium reabsorption [Peti-Peterdi et al. 2002; Komlosi et al. 2003].
Recently Ramkumar and colleagues generated mice with overexpression of renin in the collecting duct [Ramkumar et al. 2013a]. They demonstrated a fivefold increase in collecting duct renin mRNA levels in renal medulla and higher blood pressure in transgenic mice compared with control animals, suggesting that renin in the collecting duct modulates blood pressure [Ramkumar et al. 2013a, 2013b; Ramkumar and Kohan, 2013]. Further studies are necessary to establish the mechanisms by which Ang II enhances renin and (P)RR synthesis and the postranscriptional events involved.
(P)RR, cyclooxygenase 2 and RAS activation
The discovery of a receptor for renin and prorenin, (P)RR, has brought new perspectives about the possible roles of (P)RR in settings of activated intrarenal RAS. (P)RR is expressed in the kidneys, particularly in mesangial cells, podocytes and intercalated type A cells of collecting ducts [Ichihara et al. 2007, 2008; Ichihara, 2012; Gonzalez et al. 2011a]. (P)RR was described as an associated protein with V-ATPase (vacuolar H+ ATPase), giving the name to the gene ATP6AP2 [ATPase 6 accessory protein 2/(P)RR]. (P)RR binds renin and prorenin with an affinity in the nanomolar range and binding triggers a range of cellular events, like mitogen-activated kinases and extracellular signal regulated kinases (ERK 1/2). Importantly, the actions of (P)RR has been linked to diabetes nephropathy [Satofuka et al. 2009; Matavelli et al. 2010]. Despite the low levels of plasma renin in patients with diabetic nephropathy, high levels of plasma prorenin detected in these patients are associated with the occurrence of microvascular complications, microalbuminuria and retinopathy [Deinum et al. 1999]. (P)RR can activate inflammatory responses in diabetic kidneys. For example, the overexpression of (P)RR leads to augmentation of cyclooxygenase 2 (COX-2) [Kaneshiro et al. 2006]. Blockade of prorenin binding to (P)RR is able to prevent and even reverse diabetic nephropathy [Ichihara et al. 2004]. Synthesis of prorenin is also augmented in the collecting ducts in diabetic nephropathy [Kang et al. 2008], which may contribute to the activation of (P)RR and signaling pathways that promote tubular damage. Preliminary data from our group suggested that activation of (P)RR using recombinant prorenin is able to increase the expression of profibrotic genes (data not shown). However, it has been shown that intratubular Ang II can also promote tubular fibrosis [Ishidoya et al. 1995] and that collecting duct cells have the predisposition to epithelial–mesenchymal transition [Ivanova et al. 2008; Butt et al. 2007]. We demonstrated in vitro evidence that Ang II treatment of collecting duct cells stimulates fibronectin and collagen I, both markers of fibrosis, via β-catenin pathway [Cuevas et al. 2014]. Further studies are necessary to fully elucidate whether the stimulation of tubular fibrosis during intrarenal RAS activation is dependent or independent of an Ang II-mediated mechanism, that is, the direct activation of (P)RR by prorenin in the collecting duct.
Due to the demonstrations that (P)RR activation upregulates COX-2 via ERK 1/2 pathways [Kaneshiro et al. 2006] contributing to inflammatory process in the renal cortex, is it also important to evaluate what are the effects of (P)RR stimulation and overexpression in distal nephron segments and interstitial cells. We have recently reported that (P)RR and COX-2 are coexpressed in intercalated cells of the inner medullary collecting ducts [Gonzalez et al. 2013] In Figure 2(a), a colocalization of COX-2 and (P)RR in freshly isolated IMCD cells showed that both proteins coexist in the same cell type (for further characterization of specific markers please see Gonzalez and colleagues) [Gonzalez et al. 2013]. This colocalization was also observed in vivo [Figure 2(b)]. We have also shown that activation of (P)RR) using a recombinant prorenin is able to increase ERK 1/2 phosphorylation in cultured IMCD cells [Figure 3(a), modified from Gonzalez and colleagues] [Gonzalez et al. 2013]. As shown in Figure 3(b), (P)RR activation also increases COX-2 expression (modified from Gonzalez and colleagues) [Gonzalez et al. 2013]. This is important in the view that the inner medulla COX-2 metabolites play a crucial role in the maintenance of sodium/water balance and appropriate vascular tone responses during RAS activation; thus, activation of COX-2 and synthesis of COX-2 metabolites may have a role not only in inflammatory responses but also in the maintenance of the buffer mechanism against the anti-natriuretic effects of RAS activation.
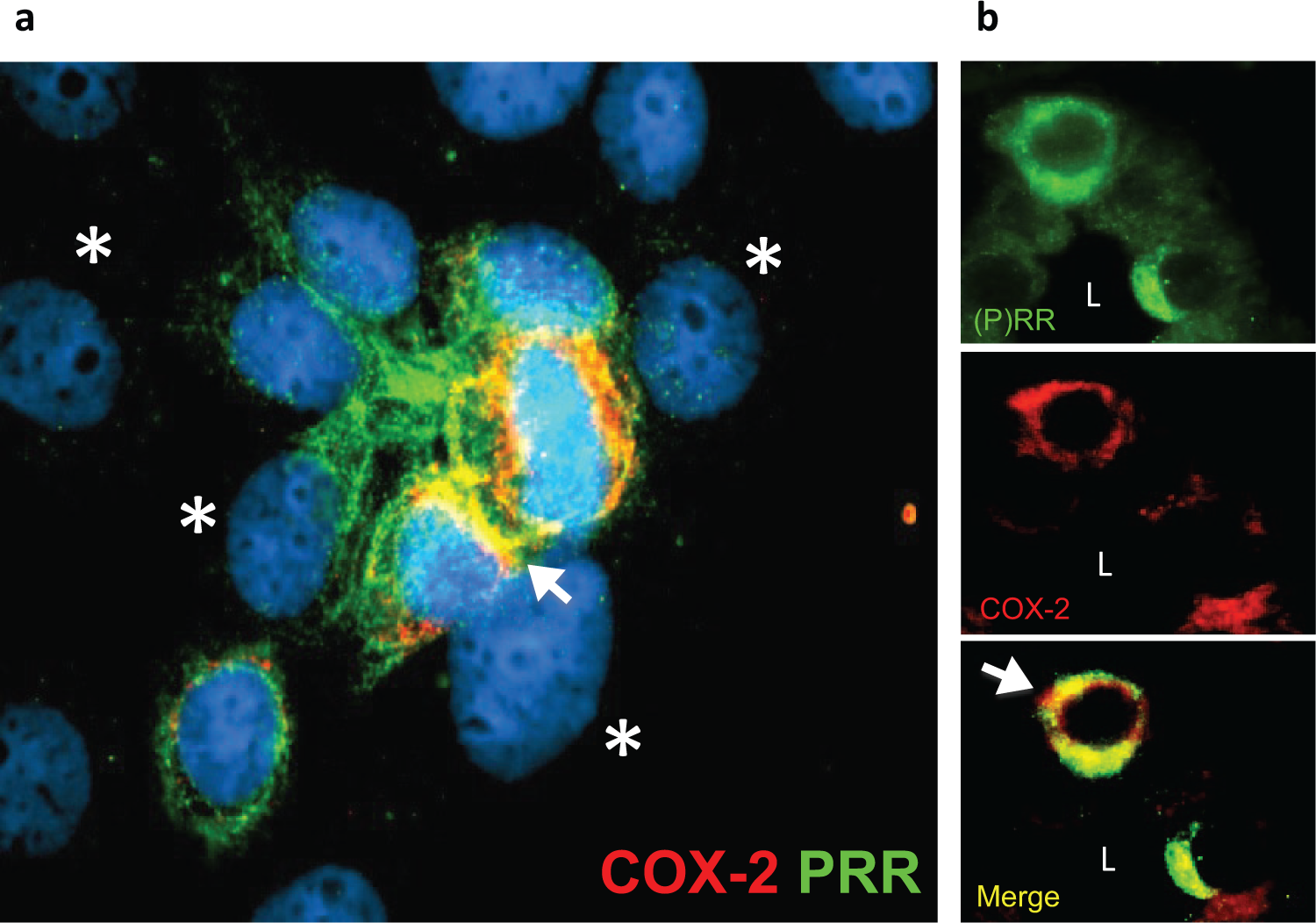
Evidence for (pro)renin receptor [(P)RR] and cyclooxygenase 2 (COX-2) expression in intercalated cells of the collecting duct. (a) Primary cultured renal inner medullary collecting duct cells stained with anti-(P)RR (green signal) and anti-COX-2 (red signal) antibodies. Yellow color (Merge) indicates co-localization in intercalated cells (arrows). Asterisks indicate principal cells which are nonstained with (P)RR antibodies. (b) Colocalization of (P)RR and COX-2 in rat kidney sections demonstrating the expression of COX-2 and (P)RR in intercalated cells (arrow). L; lumen.

Rat recombinant prorenin increases extracellular signal regulated kinase (ERK) 1/2 phosphorylation and cyclooxygenase 2 (COX-2) expression in primary cultured rat renal inner medullary collecting duct cells. (a) Time-dependent ERK1/2 phosphorylation levels reached maximal levels at 15 min. (b) COX-2 protein level was induced after 3 h of recombinant prorenin incubations and was still significant after 16 h. *p < 0.05 versus control group.
Common pathways in the regulation of (P)RR and COX-2 seem to be shared by these two proteins in renal medulla [Gonzalez et al. 2013, 2014a; Wang et al. 2014a, 2014b]. We examined the time-dependent pattern of the expression of COX-2 and (P)RR in Ang II-dependent hypertensive rats, since both are augmented in response to chronic Ang II infusions during 3, 7 and 24 days [Gonzalez et al. 2014a]. Our data showed that in the early phase of Ang II-dependent hypertension, COX-2 and (P)RR are upregulated in the inner medulla [Figure 4(a)]; this response was associated with increased ERK 1/2 phosphorylation levels [Figure 4(b)]. We did not observe this pattern at 14 days; COX-2 was not different from noninfused normotensive rats, and interestingly full length (P)RR was reduced, but soluble (P)RR was increased [Gonzalez et al. 2011a, 2014a]. Furthermore, in immunoprecipitation studies, we show renin–(P)RR interaction in hypertensive animals. Taken together, this line of evidence suggests that (P)RR activation increases COX-2 in the renal medulla. Thus, the interaction between (P)RR and renin or prorenin in plasma, interstitial space or in tubular fluids may play a physiological role. The renin and prorenin levels in the plasma are not correlated with plasma renin activity in some diseases [Deinum et al. 1999], suggesting that the final effect of the interaction between prorenin and (P)RR may depend on tissue-specific expression levels. Also, further studies are needed to elucidate the real contribution of the full-length and the soluble form of (P)RR to the increases in intrarenal Ang II generation and intracellular pathways involved in tissue damage.

Time-dependent expression of (pro)renin receptor [(P)RR] and cyclooxygenase 2 (COX-2) in renal medullary tissues of angiotensin (Ang) II infused rats. (a) Medullary protein levels of the full-length form of (P)RR and COX-2 at 3, 7 and 14 days of Ang II infusions. (b) Extracellular signal regulated kinase (ERK) 1/2 phosphorylation levels in renal medullary tissues of Ang II infused rats at 3, 7 and 14 days. *p < 0.05 versus control group.
Conclusions and perspectives
Evidence over the past 10 years of studies in this field indicates that in hypertensive models of Ang II-dependent hypertension, increased renal (P)RR transcript levels [Gonzalez et al. 2011a], as well as renin synthesis and secretion in the distal nephron segments [Prieto-Carrasquero et al. 2004, 2005, 2008; Liu et al. 2012] provide a pathway for enhanced tubular Ang II formation and the activation of intracellular pathways via (P)RR. Nguyen and associates originally reported the presence of (P)RR predominantly in glomerular mesangial cells and in vascular smooth muscle cells of renal arteries using immunofluorescence on frozen kidney tissues [Nguyen et al. 2003]. The same group described the immunoexpression of (P)RR on the basolateral side of distal tubular cells as well as in macula densa cells [Nguyen et al. 2002]. This particular cell-side localization of (P)RR might also be important for regulating Ang II levels in the renal interstitium. The increases in collecting duct renin, (P)RR gene expression and soluble form of the ℗RR(s(P)RR) activity may have a key role in mediating local augmentation of intrarenal angiotensin peptides content, since there is plenty of ACE activity in the distal nephron segments [Casarini et al. 1997; Komlosi et al. 2003; Quinto et al. 2002]. Augmented intrarenal Ang II content contributes to the pathogenesis of hypertension through sustained stimulation of Na+ reabsorption, renal vasoconstriction, and to the development of renal injury [Navar et al. 2001]. The demonstrations showing that mice infused chronically with Ang II enhance distal sodium reabsorption [Zhao and Navar, 2008; Zhao et al. 2009] emphasize further the importance that renin and (P)RR interaction may have in the distal nephron segments.
Siragy and associates showed the effects of a low-sodium diet on (P)RR expression [Matavelli et al. 2012; Huang and Siragy, 2012] in the proximal tubule. Data from our group and others also suggest that (P)RR expression is augmented during a low-sodium diet [Matavelli et al. 2012; Huang and Siragy, 2012; Gonzalez et al. 2014b]. Coincidently, with the augmented (P)RR expression in renal medulla from rats fed a low-salt diet (0.03% NaCl), renin activity in the inner medulla, as well as mRNA and protein expression are increased compared with a normal salt diet (0.4% NaCl). These data are concurrent with findings in freshly isolated rat IMCD cells grown in natural hyperosmotic conditions with preferential selectivity for IMCD principal and intercalated cells, which also exhibit increased (P)RR mRNA levels after sodium reduction (from ∼280 to ∼140 mmol/liter). Both proteins, (P)RR and renin, respond to Ang II by increasing their expression in isolated medullary collecting duct cells, indicating that Ang II regulates renin and (P)RR in the collecting duct during physiological and pathophysiological conditions.
Footnotes
Acknowledgements
The authors thank Nancy Busija, MA for the help with editing of this manuscript
Funding
Fondecyt 11121217, Chile provided support for A.A.G, the National Institutes of Health-NIDDK (DK104375-01) for M.C.P. and the Institutional Developmental Award Program of the National Center for Research Resources (P20RR-017659) for A.A.G., and M.C.P.
Conflict of interest statement
The authors declare no conflicts of interest in preparing this article.