
Abstract
The therapeutic armamentarium of parenteral anticoagulants available to clinicians is mainly composed by unfractionated heparin (UFH), low-molecular-weight heparin (LMWH), fondaparinux, recombinant hirudins (i.e. bivalirudin, desirudin, lepirudin) and argatroban. These drugs are effective and safe for prevention and/or treatment of thromboembolic diseases but they have some drawbacks. Among other inconveniences, UFH requires regular anticoagulant monitoring as a result of variability in the anticoagulant response and there is a risk of serious heparin-induced thrombocytopaenia (HIT). LMWH, fondaparinux and recombinant hirudins are mainly cleared through the kidneys and their use in patients with severe renal insufficiency may be problematic. LMWH is only partially neutralized by protamine while fondaparinux and recombinant hirudins have no specific antidote. Novel anticoagulants in development for parenteral administration include new indirect activated factor Xa (FXa) inhibitors (idrabiotaparinux, ultra-low-molecular-weight heparins [semuloparin, RO-14], new LMWH [M118]), direct FXa inhibitors (otamixaban), direct FIIa inhibitors (flovagatran sodium, pegmusirudin, NU172, HD1-22), direct FXIa inhibitors (BMS-262084, antisense oligonucleotides targeting FXIa, clavatadine), direct FIXa inhibitors (RB-006), FVIIIa inhibitors (TB-402), FVIIa/tissue factor inhibitors (tifacogin, NAPc2, PCI-27483, BMS-593214), FVa inhibitors (drotrecogin alpha activated, ART-123) and dual thrombin/FXa inhibitors (EP217609, tanogitran). These new compounds have the potential to complement established parenteral anticoagulants. In the present review, we discuss the pharmacology of new parenteral anticoagulants, the results of clinical studies, the newly planned or ongoing clinical trials with these compounds, and their potential advantages and drawbacks over existing therapies.
Keywords
Introduction
Parenteral anticoagulation may be required for the prophylaxis and/or treatment of arterial thromboembolism (ATE) or venous thromboembolism (VTE). Myocardial infarction and stroke, which are clinical manifestations of ATE, are the leading causes of mortality in developed countries [Mackman, 2008]. VTE, which encompasses deep vein thrombosis (DVT) and pulmonary embolism (PE), is the third cause of cardiovascular death in Western countries [Heit, 2008; Cohen et al. 2007a]. The therapeutic armamentarium of parenteral anticoagulants available to clinicians is mainly composed by unfractionated heparin (UFH), low-molecular-weight heparin (LMWH), fondaparinux, recombinant hirudins (i.e. bivalirudin, desirudin, lepirudin) and argatroban [Geerts et al. 2008; Hirsh et al. 2008; Kearon et al. 2008].
UFH is an anticoagulant of choice for acute coronary syndromes (ACS) [van de Werf et al. 2008], prevention of clotting during haemodialysis (HD) [Suranyi and Chow, 2010] and it remains the current standard for anticoagulation during extracorporeal circulation [Murphy and Marymont, 2007]. Major drawbacks of UFH include the variability in the anticoagulant response among patients that requires regular anticoagulant monitoring using the activated partial thromboplastin time (aPTT), risk of severe heparin-induced thrombocytopaenia (HIT), a relatively high risk of bleeding compared with other alternatives and risk of osteoporosis during chronic use [Hirsh et al. 2008; Warkentin et al. 2008].
UFH has been displaced by LMWH and fondaparinux for VTE prophylaxis and initial treatment of VTE mainly due to their longer half-life and a more predictable dose response, which allows for once-daily (OD) administration without routine anticoagulant monitoring. In addition, LMWH and fondaparinux have a reduced affinity for platelets, endothelial cells and plasma proteins. Recombinant hirudins and argatroban represent an alternative to heparin in patients with HIT [Warkentin et al. 2008] but may be also used as first-line agents in prophylaxis of VTE in patients undergoing hip or knee replacement surgery (desirudin) [European Medicines Agency, 2010c] or as an anticoagulant in patients undergoing percutaneous coronary intervention (PCI), including those patients with ACS (bivalirudin) [European Medicines Agency, 2010a]. LMWH, fondaparinux and recombinant hirudins are mainly cleared through the kidneys, and their use in patients with severe renal insufficiency may be problematic. In addition, LMWH is only partially neutralized by protamine, while fondaparinux, recombinant hirudins and argatroban have no specific antidote [Hirsh et al. 2008]. At the present time, there is no single anticoagulant strategy that is appropriate to use in all patients with acute HIT, in particular, in those undergoing cardiac surgery with extracorporeal circulation [Warkentin et al. 2008; Murphy and Marymont, 2007].
The main efforts in the anticoagulant field have been directed at the search for an oral anticoagulant capable of replacing warfarin for long-term anticoagulation (i.e. rivaroxaban, dabigatran, apixaban) [Ahrens et al. 2010; Eikelboom and Weitz, 2010; Gómez-Outes et al. 2009]. However, a number of parenteral anticoagulants are in development with the aim of complementing and, in some cases, as a substitute for the parenteral anticoagulants currently available for short-term anticoagulation. In the present review, we discuss the pharmacology of new parenteral anticoagulants, the results of clinical studies, newly planned or ongoing clinical trials with these compounds, and their potential advantages and drawbacks over existing therapies.
Mechanism of action of new parenteral anticoagulants
The new parenteral anticoagulants that are under development can block the initiation of blood coagulation in the extrinsic pathway (activated factor VII [FVIIa]/tissue factor [TF] inhibitors), may block the amplification of the coagulation cascade in the intrinsic pathway (activated factor XI [FXIa], activated factor IX [FIXa] and activated factor VIII [FVIIIa] inhibitors), or may inhibit the propagation of the coagulation (activated factor X [FXa] and activated factor V [FVa] inhibitors) and/or fibrin generation (activated factor II [FIIa] [thrombin] inhibitors) in the common pathway (Figure 1).
New parenteral anticoagulants and their targets in the coagulation cascade. Continuous line means activation while dashed line means inhibition.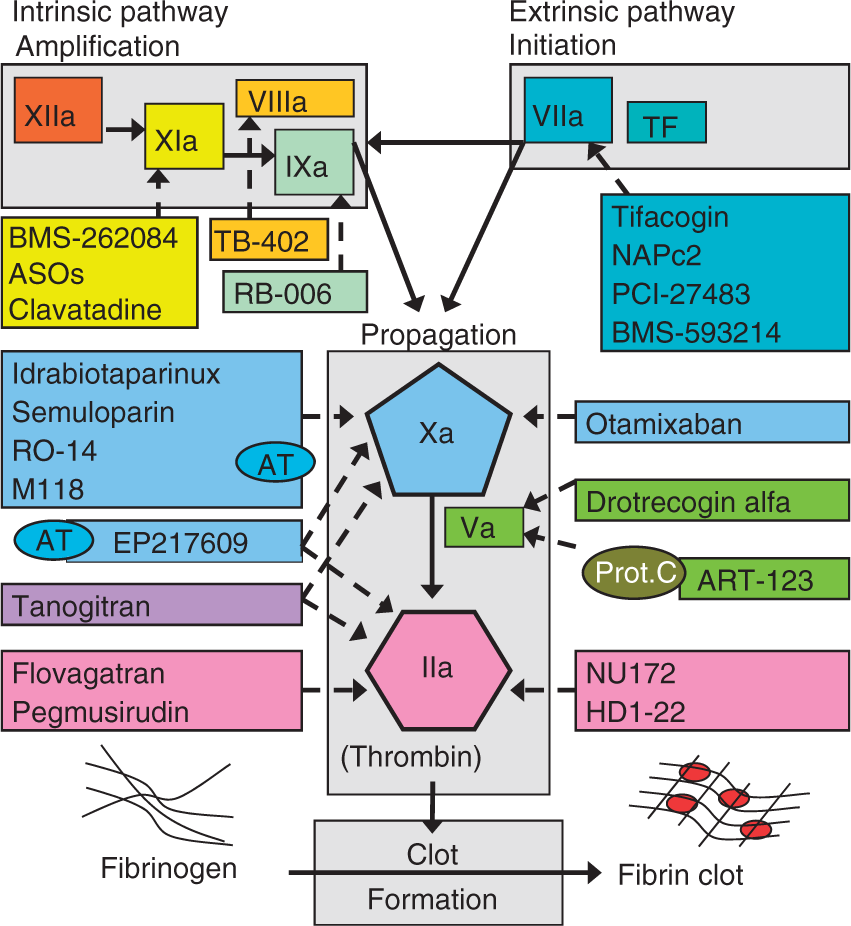
FXa and FIIa play a key role in the coagulation cascade and remain the most widely investigated targets for new anticoagulants. FXa controls thrombin generation and activation of one molecule of FX results in the generation of 1000 molecules of FIIa. Some authors have proposed that inhibition earlier in the coagulation cascade has greater antithrombotic potential [Ansell, 2007; Yin and Wessler, 1970]. Thrombin plays a central role in haemostasis by regulating blood coagulation and by inducing platelet aggregation [Davie and Kulman, 2006; Mann, 2005; Goldsack et al. 1998]. It is formed from its precursor prothrombin following tissue injury and converts fibrinogen to fibrin in the final step of the clotting cascade (Figure 1). It also promotes numerous cellular effects, including tissue repair processes, and it is involved in the pathogenesis of inflammatory and fibroproliferative disorders.
Thrombin inhibitors block the activity of thrombin whereas FXIa, FXa, FIXa, FVIIIa, FVIIa/TF and FVa inhibitors ultimately block thrombin generation and therefore may indirectly inhibit platelet aggregation. Thus, these new compounds may be effective in VTE, where fibrin formation plays an important role, and also in more platelet dominant, arterial thrombosis settings [van Ryn et al. 2007].
Indirect factor Xa inhibitors
Idraparinux sodium (SR34006; SANORG 34006) and idrabiotaparinux (biotinylated idraparinux; SSR126517E)
Idraparinux sodium (Sanofi-Aventis and Organon) is a long-acting synthetic molecule, developed based on the native pentasaccharide sequence that binds to antithrombin (AT), thus indirectly inhibiting FXa activity [Herbert et al. 1998]. It is almost completely absorbed after subcutaneous (SC) injection. The maximum concentration is reached 4 h after SC administration [Trellu et al. 2007] and it has a half-life of 120 h in healthy subjects [Ma and Fareed, 2004], allowing for once-weekly administration. It is excreted unchanged via the kidneys. Therefore, there is a risk of accumulation in patients with renal insufficiency.
A phase II dose-finding trial (PERSIST) [PERSIST Investigators, 2004], compared idraparinux (2.5, 5.0, 7.5 or 10 mg, SC, once-weekly) versus warfarin for 12 weeks in 659 patients with proximal DVT. There was no dose–response trend for efficacy, but there was a clear dose–response trend for major bleeding (0%, 3%, 1.5% and 6.9% for the 2.5, 5.0, 7.5 and 10 mg doses, respectively; p = 0.003), with an excessive major bleeding rate in patients given the 10 mg dose (6.9% vs. 0.8% warfarin). Two patients, both receiving the 5 mg dose, died due to bleeding, while patients receiving the 2.5 mg once-weekly dose had lower total bleeding rates than those who received warfarin (2.3% vs. 8.3%).
In a phase III, randomized, open-label noninferiority trial in patients with DVT (The van Gogh-DVT study, 2904 patients), idraparinux (2.5 mg once-weekly) was as effective as standard therapy (heparin followed by an adjusted-dose VKA) administered for 3–6 months [van Gogh Investigators, 2007a]. The incidence of recurrence at day 92 was 2.9% in the idraparinux group as compared with 3.0% in the standard-therapy group (odds ratio [OR], 0.98; 95% CI, 0.63–1.50), a result that satisfied the prespecified noninferiority requirement (upper limit of the 95% CI for the OR for documented symptomatic recurrent VTE of less than two). The main safety outcome, clinically relevant bleeding (CRB; the sum of major and clinically-relevant minor bleeding) rates, were 4.5% in the idraparinux group versus 7% in the standard therapy group at 3 months (p = 0.004) and 8.3% versus 8.1% at 6 months (p = 0.85). The corresponding rates of major bleeding were 0.8% versus 1.2% at 3 months (p = 0.35) and 1.9% versus 1.5% at 6 months (p = 0.50). A subgroup analysis of 421 cancer patients included in the van Gogh-DVT study showed a non-significant trend towards a lower rate of recurrent VTE at 6 months with idraparinux (2.5%) versus standard therapy (6.4%) (OR, 0.39; 95% CI, 0.14–1.11) and a similar bleeding rate (OR, 0.89, 95% CI, 0.50–1.59) and death (22.7% vs. 23.9%) [van Doormaal et al. 2010].
In patients with PE (The van Gogh-PE study, 2215 patients), the incidence of recurrence at day 92 was 3.4% in the idraparinux group (2.5 mg, once weekly) and 1.6% in the standard-therapy group (OR, 2.14; 95% CI, 1.21–3.78), with idraparinux being less effective than standard therapy [van Gogh Investigators, 2007a]. CRB rates were 5.8% in the idraparinux group versus 8.2% in the standard therapy group at 3 months (p = 0.02) and 7.7% versus 9.7% at 6 months (p = 0.10). The corresponding rates of major bleeding were 1.1% versus 2.1% at 3 months (p = 0.05) and 1.4% versus 2.8% at 6 months (p = 0.04).
During a 6-month extension of thromboprophylaxis (The van Gogh-Extension trial), idraparinux 2.5 mg once weekly was more effective than placebo in preventing recurrent VTE (1.0% vs. 3.7%; p = 0.002) but was associated with an excessive risk of a major haemorrhage (1.9% vs. 0%), including three fatal intracranial bleeds [van Gogh Investigators, 2007b].
Pharmacological properties of new direct FXa inhibitors, indirect FXa inhibitors, fondaparinux, LMWH and UFH.

LMWH, low-molecular-weight heparin; TFPI, tissue factor pathway inhibitor; UFH, unfractionated heparin.
Ardeparin, bemiparin, certoparin, dalteparin, enoxaparin, nadroparin, parnaparin, reviparin, tinzaparin.
Inhibition is achieved via binding to antithrombin.
Potential pharmacodynamic interactions with desirudin, fibrinolytic agents, GP IIb/IIIa receptor antagonists, other anticoagulants, antiplatelet drugs (acetylsalicylic acid, dipyridamole, sulfinpyrazone, ticlopidine or clopidogrel), and nonsteroidal anti-inflammatory drugs.
Partial neutralization.
Idrabiotaparinux (biotinylated idraparinux) (Sanofi-Aventis) is a long-acting synthetic pentasaccharide. It displays similar anticoagulant properties than idraparinux [Savi et al. 2008]. A differential characteristic is that, due to binding to biotin moiety, the anticoagulant effect of idrabiotaparinux can be rapidly neutralized following the intravenous (IV) infusion of avidin (EP5001), an egg-derived protein [Paty et al. 2010].
The EQUINOX study compared the efficacy and safety of idrabiotaparinux 3 mg and idraparinux 2.5 mg in the treatment of acute DVT [Buller et al. 2008]. Rates of recurrent VTE and of fatal or nonfatal PE were similar with idrabiotaparinux (2.3% [9 of 385 patients]) and idraparinux (3.2% [12 of 370 patients]) at 6 months. There was less clinically relevant bleeding (5.2% versus 7.3%) and less major bleeding (0.8% versus 3.8%) with idrabiotaparinux than with idraparinux. The finding of lower bleeding rates with idrabiotaparinux (before antidote administration) than with idraparinux is intriguing, given that both drugs are supposed to have similar anticoagulant properties. At the end of the 6-month study period, 52 idrabiotaparinux patients were re-randomized to receive 30-minute IV infusion of avidin 100 mg or placebo. Avidin infusion after the last idrabiotaparinux injection led to rapid, substantial and maintained reversal of anti-Xa activity and was well-tolerated [Buller et al. 2008].
Idrabiotaparinux was tested in comparison with warfarin for stroke prevention in AF (BOREALIS-AF study, 9600 patients) [ClinicalTrials.gov, NCT00580216], but the study was stopped and the development in this indication has been discontinued. Considering recent therapeutic advances in this field, this compound does not appear able to bring significant improvement in the care of these patients (Sanofi-Aventis Press release, 21 December 2009, www.sanofi-aventis.com). There is an ongoing study aimed at showing noninferiority of this new compound versus standard treatment for acute symptomatic PE (CASSIOPEA study, 3200 patients) [ClinicalTrials.gov, NCT00345618]. The estimated completion date is October 2010.
Ultra-low-molecular-weight heparins
Semuloparin sodium (AVE5026; Sanofi-Aventis) is a new ultra-low-molecular-weight heparin (ULMWH) (mean molecular weight [MW]: 2400 Da), with predominantly anti-FXa activity (ratio anti-FXa/anti-FIIa >30) [Viskov et al. 2009] and a half-life between 11-22 h [Dubruc et al. 2009], which allows OD SC administration. It is obtained by phosphazene promoted depolymerization of heparin from porcine intestinal mucosa, leading to a pool of polysaccharide chains with an individual MW distribution. No more than 40% is inferior to 1600 Da and no more than 11% is superior to 4500 Da [World Health Organization, 2009]. A low mean MW and a high anti-Xa/IIa activity ratio might be associated with a high antithrombotic efficacy with reduced bleeding tendency [Ansell, 2007]. In human plasma, potent inhibition of thrombin generation by AVE5026 was closely related to its anti-FXa activity. In a rat venous thrombosis model, AVE5026 showed a dose-dependent antithrombotic activity comparable to that of enoxaparin (median effective dose [ED50]: AVE5026 = 1.6 mg/kg and enoxaparin = 2.8 mg/kg). Similar results were obtained in rabbits and dogs. At equipotent doses, AVE5026 did not affect bleeding parameters, whereas enoxaparin showed increased haemorrhage in rats, rabbits and dogs [Viskov et al. 2009]. The anticoagulant effects of AVE5026 are not neutralized by protamine [Eikelboom and Weitz, 2010]. Preliminary in vitro evaluation of immune-mediated HIT indicates a low risk for AVE5026 [Viskov et al. 2009].
Pharmacokinetic (PK) data from the first three phase I studies [Dubruc et al. 2009] included 143 subjects. After a single SC 60 mg dose in 15 subjects, bioavailability was 98% compared with IV administration, the total clearance was 0.600 ± 0.173 l/h and the volume of distribution (VD) was 7.88 ± 1.45 l. After a single dose in 56 subjects, anti-Xa concentration increased in proportion to the dose from 0.1 to 1.4 mg/kg. In the repeated dose-escalation study, data from 72 subjects were available for the anti-Xa PK analysis. After repeat dosing from 20 to 100 mg in elderly subjects, a slight increase in accumulation of AVE5026 by 1.47-fold from day 1 to day 14 was seen. Half-life ranged from 11 hours after single doses in young volunteers to 22 hours after repeated doses in elderly subjects. Overall, AVE5026 was well tolerated and safe [Dubruc et al. 2009].
The phase IIb TREK study [Lassen et al. 2009], which involved 690 randomized patients undergoing total knee replacement (TKR) surgery, investigated the dose–response trend of AVE5026. The study showed a highly significant dose–response trend (p < 0.0001) on the primary efficacy end point (total VTE plus VTE-related deaths until post-operative day 11) with event rates of 40%, 44.1%, 15.6%, 13.6% and 5.3% for the AVE5026 SC doses of 5, 10, 20, 40 and 60 mg, respectively. The VTE rate in the enoxaparin 40 mg arm was 35.8%. A statistically significant dose response (p = 0.0003) was found for total bleeding with rates, ranging from 3.8% to 20.5% for the AVE5026 dosing arms, compared with 5% in the enoxaparin arm.
Phase III studies with semuloparin 20 mg OD for VTE prophylaxis.

SAVE-KNEE: Elective Knee Replacement Surgery; SAVE-HIP1: Total Hip Replacement Surgery; SAVE-HIP2: Hip Fracture Surgery; Meta-analysis: meta-analysis of three studies (SAVE-KNEE, SAVE-HIP1 and SAVE-HIP-2); SAVE-HIP3: Hip fracture surgery (extended prophylaxis); SAVE-ABDO: Major abdominal surgery; SAVE-ONCO: Medical patients undergoing chemotherapy; SAVE-VEMED: Acutely ill medical patients with restricted mobility; BID; twice daily; OD, once daily; ENO, enoxaparin; VTE, venous thromboembolism; OR, odds ratio; CI, confidence interval; NA, not available.
Total VTE is the sum of proximal and distal DVT, either symptomatic or asymptomatic detected by bilateral elective ascending contrast venography plus symptomatic PE documented by objective methods.
Clinically relevant bleeding is the sum of major bleeding and any clinically relevant nonmajor bleeding.
Terminated after 421 patients had been enrolled (initially planned: 12,300).
RO-14 (Rovi Pharmaceuticals Laboratories) is a new ULMWH (MW ≈ 2500 Da), with predominantly anti-Xa activity and virtually devoid anti-IIa activity. In healthy volunteers, ascending SC doses of RO-14 (from 1750 to 19,950 anti-Xa IU) showed a proportional and linear PK. Elimination half-life was 6.8 h. No clinically significant changes were observed in aPTT and TT and all RO-14 doses were well tolerated [Antonijoan et al. 2009].
New low-molecular-weight heparins
M118 (Momenta Pharmaceuticals) is a novel LMWH (mean MW: 6500 Da) with predominantly anti-FXa activity (ratio anti-FXa/anti-FIIa = 1.4) which is produced by depolymerization of UFH that is derived from porcine intestinal mucosa [Kishimoto et al. 2009]. It has been designed specifically for use in the treatment of ACS. M118 shows potent activity against both FXa (≈240 IU/mg) and thrombin (FIIa; ≈170 IU/mg). M118 achieved superior activity to conventional LMWH in a rabbit model of abdominal arterial thrombosis without increasing bleeding risk [Kishimoto et al. 2009]. SC bioavailability in humans is around 70%. Half-life is approximately 1 hour after IV bolus injection and 2–3 h after SC injection (Table 1). In addition, M118 seems not to activate platelets, its anticoagulant activity is reversible with protamine sulfate (1 mg per 100 IU dose) and monitorable by standard coagulation assays [Rao et al. 2010].
The EMINENCE phase IIb, multicentre study [Rao et al. 2010; Melloni et al. 2009] included 503 patients undergoing PCI that were randomized in an open-label fashion to one of three M118 IV dosing arms (50, 75 or 100 IU/kg) or UFH 70 IU/kg as calibrator. The primary end point (composite of death, myocardial infarction, repeat revascularization, stroke, thrombocytopenia, catheter thrombus, bailout use of glycoprotein IIb/IIIa inhibitor, or any bleeding through 30 days) occurred in 22.7%, 28.3% and 30.1% of patients randomized to the M118 doses of 50, 75 and 100 IU/kg, respectively, and in 31.1% of patients randomized to UFH. The adverse event profiles of M118 and UFH were comparable. This study showed that M118 is well tolerated and feasible to use as an anticoagulant in patients undergoing elective PCI. Further confirmatory studies are needed.
Low-molecular-weight lignins
Chemo-enzymatically prepared sulfated low-molecular-weight lignins (LMWLs) represent a library of diverse nonsugar, aromatic molecules with structures radically different from the heparins that mimic the biological activities of heparin and heparan sulfate [Henry et al. 2010, 2009a, 2009b, 2007; Monien et al. 2006]. Competitive binding studies show that the sulfated LMWLs bind to the pentasaccharide-binding site and extended heparin-binding site of antithrombin [Henry et al. 2009a], but also may interact with exosite II of thrombin [Henry et al. 2007]. LMWLs have been found to inhibit FXa and thrombin, and to prolong prothrombin time (PT) and aPTT at concentrations similar to LMWH [Henry et al. 2009b; Monien et al. 2006]. In addition, LMWLs may inhibit plasmin with IC50 values in the range of 0.24 and 1.3 µM [Henry et al. 2010]. One designed sulfated LMWL specifically inhibits thrombin with an IC50 value of 10–20 nM [Desai, 2009/2010]. Studies using A549 lung and HepG2 liver cell lines showed no toxicity at concentrations as high as 50 mg/l. These agents are currently in the preclinical phase.
Direct factor Xa inhibitors
Otamixaban (XRP0673, FXV673, RPR130673)
Otamixaban (Sanofi-Aventis) is a synthetic, potent (Ki = 0.5 nM), highly selective direct FXa inhibitor that is administered IV and has a short half-life (approximately 30 min after IV administration) (Table 1) and quick onset/offset of action [Hinder et al. 2006a]. In vivo experiments showed that otamixaban is effective in rodent, canine and porcine models of thrombosis. The major route of elimination of otamixaban is biliary (71%; as the reduced metabolite MA, 35) and urine (25%; as otamixaban). Reductive metabolism presumably occurs in the gastrointestinal tract [Guertin and Choi, 2007].
An otamixaban dose ranging phase I study (80 healthy male volunteers; 60 drug treated, 20 placebo) was designed to assess the safety, PK and specific PD markers of anti-coagulation [Paccaly et al. 2005]. Overall, otamixaban was well tolerated over the 100-fold dosing regimen (from 1.7 to 183 µg/kg/h, 6-hour infusion). The predicted target antithrombotic and therapeutic otamixaban plasma concentration (100 ng/ml) was achieved after a 53 µg/kg/h, 6-hour infusion. At this dose both aPTT and PT did not change markedly (1.2-fold increase) however the Heptest Clotting Time (HCT) appeared to be a more sensitive marker (2.7-fold increase). In healthy male volunteers, no significant drug–drug interaction was observed on the coagulation or platelet aggregation with the co-administration of otamixaban with tirofiban [Hinder et al. 2005] or ASA [Hinder et al. 2006b].
In a randomized, placebo-controlled, double-blind, multicentre study to assess preliminary safety, PK and PD of otamixaban in 119 patients with stable coronary artery disease (CAD), otamixaban exerted a rapid onset of coagulation, with anti-FXa activity and anticoagulant effect being measurable 3 minutes after the start of otamixaban administration (1-minute bolus followed by a 24-hour continuous infusion) [Hinder et al. 2006a]. Upon cessation, these effects declined rapidly and returned to baseline within 6 hours after the end of infusion. Fifty percent of the patients had mild renal dysfunction. However, renal impairment did not affect the systemic clearance of otamixaban.
The SEPIA-PCI was a phase II double-blind, double-dummy, parallel-group, dose-ranging trial that compared five different weight-adjusted otamixaban regimens (bolus plus IV infusion: from 0.025 mg/kg followed by 0.035 mg/kg/h [dose 1] to 0.140 mg/kg followed by 0.200 mg/kg/h [dose 5]) and weight-adjusted UFH in 947 patients before PCI [Cohen et al. 2007b]. The median change in prothrombin fragments 1 + 2 (F1 + 2) from baseline to the end of infusion (primary end point) was greater with the highest otamixaban dose compared with UFH (−0.3 versus −0.2 ng/ml; p = 0.008). Anti-FXa levels ranged from 65 to 691 ng/ml with otamixaban doses 1 and 5, respectively. Significant TIMI bleeding (main secondary end point) ranged between 1.9% and 3.9% with different otamixaban regimes and 3.8% for UFH. Ischaemic events (a secondary end point) ranged between 3.8% and 7.1% in different otamixaban regimes and 5.6% for UFH.
The SEPIA-ACS1 TIMI 42 trial [Sabatine et al. 2009] was a double-blind, phase IIb trial undertaken in 3241 patients with non-ST-elevation ACS that compared five doses of otamixaban (0.08 mg/kg bolus followed by infusions of 0.035 [n = 125], 0.070 [n = 676], 0.105 [n = 662], 0.140 [n = 658] or 0.175 [n = 671] mg/kg/h) versus UFH (60 IU/kg IV bolus followed by an infusion of 12 IU/kg/h) plus eptifibatide (180 µg/kg IV bolus followed by an infusion of 1.0–2.0 µg/kg/min) (n = 449). Enrolment into the lowest dose group was stopped early due to increased thrombotic events. Rates of the primary efficacy end point (a composite of death, myocardial infarction, urgent revascularization, or bailout glycoprotein IIb/IIIa inhibitor use up to 7 days) in the five otamixaban doses were 7.2% with 0.035 mg/kg/h, 4.6% with 0.070 mg/kg/h, 3.8% with 0.105 mg/kg/h, 3.6% with 0.140 mg/kg/h and 4.3% with 0.175 mg/kg/h (p = 0.34 for trend). In the control group, the rate was 6.2%. Thrombotic complications during PCI were numerically higher with the two lower otamixaban doses than with UFH plus eptifibatide (RR for otamixaban doses 0.035 and 0.070 mg/kg/h combined versus UFH + eptifibatide was 2.13; 95% CI, 0.94–4.85; p = 0.06). Rates of the primary safety end point (TIMI major or minor bleeding not related to coronary-artery bypass grafting) in the five otamixaban doses were 1.6%, 1.6%, 3.1%, 3.4% and 5.4%, respectively (p = 0.0001 for trend). In the control group the rate was 2.7%. On the basis of these results, the authors proposed that intermediate doses of otamixaban (0.105 and/or 0.140 mg/kg/h) may be suitable for further testing in a phase III confirmatory trial.
Otamixaban is being tested for superiority against UFH + eptifibatide in the ongoing phase III TAO study in 10930 patients with unstable angina/non-ST segment elevation myocardial infarction scheduled to undergo an early invasive strategy [ClinicalTrials.gov, NCT01076764]. The main efficacy end point will be the composite of all-cause death, myocardial infarction and any stroke. The results are expected in June 2012.
Direct thrombin inhibitors
Pegmusirudin (PEG-hirudin; SPP200; LU87981)
Pegmusirudin (PEG-hirudin, Canyon Pharmaceuticals; SPP200, licensed from Speedel; LU87981, licensed from Abbott), a highly selective direct thrombin inhibitor, is a chemically defined conjugate of recombinant hirudin (r-hirudin) and two molecules of polyethylene glycol (PEG)-5000 with a mean molecular mass of 17 kDa [Avgerinos et al. 2001; Pöschel et al. 2000; Humphries et al. 1997]. In healthy subjects, its elimination half-life ranged from 18 to 24 h [Esslinger et al. 1997], while in patients with severe renal insufficiency but not undergoing HD, its elimination half-life was further prolonged to 38.4 h [Pöschel et al. 2000].
A preliminary study was conducted in 20 patients with end-stage renal disease to assess the appropriate pegmusirudin dosage regimen in patients on HD, as well as to compare the safety, tolerability and efficacy of the compound with that of UFH [Pöschel et al. 2004]. Dialysis sessions lasting a mean of 4.3 hours were performed three times a week. The starting dose of PEG-Hirudin in most patients was a single 80 µg/kg bolus injection (HD4). Before each of the following sessions (HD5–13), an individualized PEG-Hirudin dose of between 26 and 65 µg/kg (mean dose 41 µg/kg) was injected. Five minutes after injecting PEG-Hirudin or UFH, mean aPTT was prolonged to a maximum of 85 and 188 seconds, respectively. Mean post-dialysis aPTT values ranged from 60–68 seconds after PEG-Hirudin and 34 to 46 seconds after UFH. Mean predialysis pegmusirudin plasma concentrations ranged between 488 and 536 ng/ml. Mean predialysis aPTT was prolonged by 46–56 seconds by PEG-Hirudin but not affected by UFH, indicating an anticoagulant effect of PEG-Hirudin between dialysis sessions.
A further phase II trial compared PEG-Hirudin (initial loading dose of 0.08 mg/kg [HD1], followed by a 3-month period [HD2–42] of either 0.04 mg/kg or 0.05 mg/kg to maintain the aPTT ratio between 1.5 and 2.5) versus UFH in 127 patients on HD [Mann et al. 2006]. After 3 months, the PEG-Hirudin dose was uptitrated by 30% (0.052 or 0.065 mg/kg) for another 3 months (HD43–84). The frequency of vascular graft occlusions (VGOs) was significantly lower with PEG-Hirudin than with UFH (11.1% vs. 40.5% between HD1 and HD84; p < 0.001). Major bleeding events were more frequent in the PEG-Hirudin group (8.4%) compared with UFH (5.4%). There were also more minor bleeds with PEG-Hirudin than UFH (57.8% vs. 32.4% between HD1 and HD84; p < 0.001). The aPTT did not allow the bleeds to be predicted. In summary, although pegmusirudin appears to be an effective new anticoagulant therapy to prevent clotting in HD, the higher rate of bleeding events compared with UFH and the lack of specific antidote have limited further development of the drug in this indication. Pegmusirudin is currently in preclinical development in cancer and cancer-related thrombosis [Esser et al. 2009].
Flovagatran sodium (TGN 255)
Flovagatran sodium (Paion, licensed from Trigen) is a potent (Ki = 7–22 nM), reversible, low MW (565.5 Da), highly selective synthetic direct thrombin inhibitor that is administered IV [Wang et al. 2007; Chahwala and Chander, 2005; Combe et al. 2005a].
In healthy volunteers, flovagatran showed dose linearity with a good systemic exposure and distribution (VD ≈ 40 l). A 30-second bolus induced a rapid (within 2 min) increase in plasma concentration of the drug. A steady state was achieved within 4–6 h after the start of a 3-hour infusion. The disposition of TGN 255 followed a biphasic pattern with a short distribution phase with a half-life of 6–8 min, and an effective systemic half-life of 1.7–2.3 h. Approximately 14% of the administered dose was excreted as TGN 255 [Combe et al. 2005a]. Infusion of TGN 255 induced a rapid, dose-related increase in the thrombin time (TT), reaching a peak 2–3 h after the start of the infusion (maximum 4.5- to 7.5-fold higher than at predose). Following cessation of the infusion there was a biphasic fall in TT to threefold baseline values within 30 min. There was a linear correlation between ecarin clotting time (ECT) and TT, as well as between ECT and plasma concentration of TGN 255 [Combe et al. 2005b]. A phase I interaction study between flovagatran (80 mg/h, as IV infusion over 6 h) and aspirin 300 mg plus clopidogrel 150 mg showed no differences in TT, aPTT and PT time whether flovagatran was administered alone or on top of aspirin plus clopidogrel [Combe et al. 2006a].
A small phase IIa open-label multicentre trial was conducted to assess TGN 255 as anticoagulation of the extracorporeal circuit in patients undergoing chronic HD [Combe et al. 2006b]. A total of 84 sessions in 27 patients were analysed. Increased doses of TGN 255 were associated with decreased extracorporeal circuit clotting such that at final rates greater than 25 mg/h no clotting was reported. Activated whole blood clotting time (AWBCT) increased by 50% and remained stable for the rest of the observation period. TT rapidly increased after start of TGN 255 and decreased rapidly after cessation of TGN 255 infusion. A less marked change was observed for aPTT. No haemorrhage from any site was reported in any patient; one serious hyperkalaemia was reported in one patient on heparin.
TGN 255 provided effective anticoagulation in a canine cardiopulmonary bypass (CPB) procedure, enabling successful completion with minimal blood loss [Nelson et al. 2008]. These findings support further evaluation of TGN 255 as an anticoagulant for CPB.
NU172
NU172 (ARCA biopharma &Nuvelo) is a direct antithrombin DNA aptamer (term derived from the Latin ‘aptus’, to fit; a protein-binding oligonucleotide) [Wagner-Whyte et al. 2007]. NU172 was discovered within a degenerate DNA oligonucleotide library using SELEX (systematic evolution of ligands by exponential enrichment) and was subsequently truncated to 26 nucleotides [Keefe et al. 2010]. It has an IC50 value of 5–10 µg/ml in plasma in an ECT assay as measured by thromboelastography [Waters et al. 2009]. Because NU172 is not capped, substituted or conjugated to PEG, it has a short duration of action in vivo [Hutabarat et al. 2007].
In a phase Ia study in 30 healthy volunteers, NU172 produced dose-dependent increases in activated clotting time (ACT). Upon withdrawal of NU172 the ACT showed a rapid return toward baseline with a plasma half-life of NU172 of approximately 10 minutes. No serious adverse events occurred in the trial (Nuvelo press release, 29 April 2008, www.nuvelo.com). A further phase 1b study testing in 24 healthy male volunteers who received a 2 mg/kg bolus, followed by escalating infusion doses for 4 h, was carried out. The highest infusion dose rate tested, 6 mg/kg/h, resulted in an average ACT increase of approximately three times baseline, and an average PT and aPTT increase of approximately five times baseline. All measurements were maintained stably throughout the 4-hour infusion. Once the infusion ended, the ACT and other coagulation parameters showed a rapid return toward baseline. No serious adverse events were reported (Nuvelo press release, 14 August 2008, www.nuvelo.com).
Owing to their PK/PD characteristics (parenteral administration, short half-life, rapid onset–offset of action, potent anticoagulant activity), NU172 is a good candidate to be further investigated as anticoagulant during invasive and/or surgical cardiovascular procedures. A phase II clinical trial is planned to evaluate NU172 as anticoagulation in 30 patients undergoing off-pump CABG surgery (SNAP-CABG-OFF) [ClinicalTrials.gov, NCT00808964].
HD1-22 and HD1
HD1-22 (LIMES Institute) is a bivalent aptamer that binds to thrombin with high affinity (Kd = 0.65 nM) and occupies both anion binding exosites without blocking the active centre of the enzyme [Müller et al. 2008, 2007]. One of their components, the univalent HD1 DNA aptamer, has shown potent anticoagulant properties that reflect its capacity to attenuate FV activation by thrombin and inhibit prothrombinase assembly [Kretz et al. 2010]. HD1-22 has been developed by connecting the exosite 1 binding aptamer HD1 and the exosite 2 binding aptamer HD22 through a poly-dA linker. HD1-22 prolongs clotting times of the TT, aPTT, ECT and lag time of the TF triggered thrombin generation assay in a dose-dependent manner. On a molar basis, its anticoagulant activity is nearly identical to bivalirudin and superior to argatroban. The anticoagulant activities of HD1-22 were fully reversed by addition of the antidote-oligodeoxynucleotide AD1-22/59 [Müller et al. 2008]. HD1-22 is therefore an interesting anticoagulant candidate for use in clinical situations where effective anticoagulation and rapid reversal of the anticoagulant effect are required. The data obtained warrant further studies.
Factor XIa inhibitors
Targeting proteases upstream of the final common pathway of coagulation may provide anticoagulation without impeding haemostasis. This concept is supported by observations in human and mouse FXI deficiency, with large- and small-molecule inhibitors of FXI activity [Schumacher et al. 2010]. Some of these compounds are in preclinical testing.
BMS-262084
BMS-262084 (Bristol-Myers Squibb) is an irreversible and selective small-molecule inhibitor of FXIa with an IC50 of 2.8 nM against FXIa [Schumacher et al. 2007]. BMS-262084 also inhibits tryptase and earlier studies of allergic bronchoconstriction and airways inflammation showed protective effects of aerosol-administered BMS-262084 [Sutton et al. 2002]. In rat models of thrombosis and haemostasis, BMS-262084 doubled the aPTT in human and rat plasma at 0.14 and 2.2 µM, respectively, while PT was unaffected [Schumacher et al. 2007]. BMS-262084 administered as an IV loading plus sustaining infusion was effective against FeCl(2)-induced thrombosis in both the vena cava and carotid artery. A recent in vitro study showed that BMS-262084 did not prevent TF-induced platelet aggregation, whereas inhibition of thrombin generation by blocking upstream proteases (FVIIa inhibition with BMS-593214 and FXa inhibition with rivaroxaban) in the blood coagulation cascade was as effective as direct thrombin inhibition with dabigatran in preventing TF-induced platelet aggregation [Wong and Jiang, 2010].
Antisense oligonucleotides
Antisense oligonucleotides (ASOs) are novel therapeutic agents that inhibits gene by cleaving specific mRNA sequences using hybridization and RNase H [Keefe et al. 2010]. ASOs need to cross the cell membrane to reach their target. Subsequent selective cleavage of the target mRNA leads to a corresponding reduction in target protein. Initial preclinical studies were conducted with FII and FIX ASOs. Inhibition of the corresponding target mRNAs were able to reduce thrombosis, but had deleterious effects on the bleeding tendency [Crosby et al. 2009; Monia et al. 2007]. Selective second-generation ASOs targeting FXI have been developed and characterized for their efficacy and safety in various murine models of thrombosis and bleeding [Zhang et al. 2010]. Systemic treatment of mice with an FXI ASO, called ISIS 404071, led to specific, potent and dose-dependent reduction of hepatic FXI mRNA levels with corresponding reductions in plasma levels of FXI protein and activity [Zhang et al. 2010]. Similar results were obtained with FXI antisense oligonucleotides called FXI-AS1 and FXI-AS2 after SC administration in cynomolgus monkeys [MacLeod et al. 2009]. FXI antisense treatment produced potent and dose-dependent antithrombotic activity in various venous and arterial thrombosis models, comparable to warfarin or enoxaparin. Co-administration of FXI ASO with enoxaparin or the antiplatelet drug clopidogrel produced improved antithrombotic activity without increased bleeding [Zhang et al. 2010]. Finally, plasma-derived FXI concentrate was shown to effectively and rapidly reverse the anticoagulant effect of FXI antisense therapy [Zhang et al. 2010]. Therefore, inhibition of FXI through ASOs might serve as a new strategy for the treatment and prevention of thromboembolic disease. Further evaluation in humans is warranted.
Clavatadine
Clavatadine A and B are natural products isolated from a marine sponge. They inhibit FXIa by irreversible covalent bonding of the carbamate functional group to the active serine residue in the blood-clotting factor with IC50s of 1.3 and 27 µM, respectively [Buchanan et al. 2008]. There is a project to synthesize clavatadine A and to change the covalent bonding of the carbamate group to make the molecule have reversible activity, thus making it safer for human use [Vreeland, 2010].
Factor IXa inhibitors
Coagulation FIXa is primarily associated with the intrinsic and propagation phase of coagulation. It plays a pivotal role in TF-mediated thrombin generation thus representing an attractive target for anticoagulant development [Eikelboom et al. 2010]. Two FIXa inhibitors have undergone testing in phase I or II trials (oral TTP889 and parenteral RB006). The latter is discussed here.
RB006
RB006 (Regado Biosciences Inc.) is an RNA aptamer that reversibly inhibits FIXa with high affinity and specificity [Becker et al. 2010; Rusconi et al. 2002]. By targeting FIXa, RB006 has the potential to inhibit the activation of coagulation induced by exposure of blood to artificial surfaces, such as stents or cardiac bypass circuits. RB006 is composed by 34 nucleotides, conjugated to 40-kDa PEG to reduce its renal filtration thus increasing half-life and capped with an inverted nucleotide at the 3’-terminus to reduce 3’-exonuclease-mediated degradation [Keefe et al. 2010]. Clearance of free RB006 involves both intravascular and to a lesser degree renal mechanisms. Unlike functional nucleic acids (i.e. antisense oligonucleotides), the current generation of aptamers do not exert their effects intracellularly [Becker et al. 2010]. In vitro, RB006 inhibits thrombin generation and clot formation in a concentration-dependent manner [Tanaka et al. 2009]. RB006 is being developed with its complementary oligonucleotide antidote (RB007) as the anticoagulation systems called REG1 (IV RB006 and IV RB007) and REG2 (SC RB006 and IV RB007) (Regado Biosciences). The RB006-RB007 complex is both stable and biologically inactive. Its clearance is believed to involve metabolism of the active agents to inactive nucleotides by endogenous endonucleases. Given the rapid clearance of RB007 and the RB006-RB007 complex, re-anticoagulation can be achieved within minutes of RB007 administration by administering RB006 [Becker et al. 2010]. As an aptamer/antidote pair, RB006/RB007 has the potential to be an alternative to heparin/protamine in cardiac surgery.
In a phase Ia study in healthy volunteers [Dyke et al. 2006] and phase Ib study in patients with stable CAD [Chan et al. 2008a], a predictable dose–response trend, reflected in aPTT measurements, was seen after administration of the bolus of RB006. Full recovery of coagulation after administration of IV RB007 to neutralize IV RB006 was found to be both rapid (1–5 minutes) and durable (up to 7 days) [Chan et al. 2008a]. In an additional PK study, 39 healthy volunteers were randomised to receive either three consecutive weight-adjusted, drug-antidote treatment cycles, or double placebo [Chan et al. 2008b]. Each treatment cycle included an IV bolus of 0.75 mg/kg RB006, followed 60 minutes later by a descending dose of RB007, ranging from a 2 : 1 to 0.125 : 1 antidote/drug ratio (1.5 to 0.094 mg/kg RB007). Repeat doses of RB006 achieved highly reproducible aPTT levels with low intrasubject variability, while repeat doses of RB007 reversed the aPTT levels dose-dependently. There was no major bleeding or serious adverse events.
REVERSAL PCI was a phase IIa randomized trial comparing REG1 (n = 20) with UFH (n = 4) in elective PCI [Cohen et al. 2010]. RB006 was given as a 1 mg/kg IV bolus. Coagulation measures at 5 min post-RB006 dosing and at the end of PCI were: plasma aPTT 2.5-fold and 2.4-fold above baseline; whole blood aPTT 148.5 and 145 seconds; ACT 223 and 236 seconds. In the respective assays, partial reversal with a 50% reversal dose of RB007 resulted in 51–68% reversal of RB006, while complete reversal RB007 resulted in 93–103% reversal of RB006. All PCI procedures were successful, with minimal bleeding and no intraprocedural thrombotic events. A randomized phase IIb dose-finding study assessing the efficacy and safety of REG-1 system compared with heparin in approximately 800 patients with ACS undergoing PCI (RADAR study) is currently ongoing [ClinicalTrials.gov, NCT00932100]. The estimated study completion date is December 2010.
The REG-2 system composed by the SC administration of RB006 paired with IV administration of its antidote RB007 was first tested in two studies in monkeys [Rusconi et al. 2010]. Single RB006 SC doses resulted in peak anticoagulation at 12 h, which persisted until at least 48 h and decayed over 7 days. Administration of RB007 at the time of RB006 peak effect neutralized the absorbed RB006 but did not further affect absorption of RB006 remaining at the site of injection. A phase I, partially blinded, five-arm, single ascending dose study with the REG-2 system was conducted in 36 healthy volunteers [Zelenkofske et al. 2010]. SC RB006 was administered abdominally as a one-time injection. Each dose was escalated after safety evaluation (Groups 1, 1a: 0.5 mg/kg; Group 2: 1 mg/kg; Group 3: 3 mg/kg). A fifth cohort received planned reversal with RB007. Compared with IV RB006, SC RB006 exhibited a delayed plasma appearance (mean 2.5 h), delayed Tmax (mean 72 h) and a longer half-life (6–7 days), but was in a similar plasma concentration range. Dose-dependent aPTT prolongations were consistent with plasma levels. Reversal of plasma RB006 with RB007 was immediate with continued absorption of residual SC RB006 and re-anticoagulation to a lesser extent over time. The injections were well tolerated. Further studies are needed to test clinical utility.
Factor VIIIa inhibitors
FVIIIa is associated with the intrinsic and propagation phase of coagulation. It is a cofactor for FIXa, which in the presence of calcium and phospholipids forms a complex that activates FX, thus representing a potential target for new anticoagulants.
TB-402 (Mab-LE2E9Q)
TB-402 (ThromboGenics) is a human monoclonal antibody (fully human IgG4 antibody) that partially inhibits FVIIIa [Verhamme et al. 2010; Jacquemin et al. 2006]. That antibody was generated by introducing a point mutation in the gene of an anti-FVIII antibody, Mab-LE2E9 [Jacquemin et al. 2000]. As a result, Mab-LE2E9Q inhibits only about 40% of FVIII activity and does not prevent FVIII from binding to VWF [Jacquemin et al. 2006]. Preclinical studies confirmed this plateau inhibition and the antithrombotic effects of TB-402 [Jacquemin et al. 2009, 2006; Dewerchin et al. 2004; Singh et al. 2002]. Its half-life (±3 weeks) may allow for prolonged prophylaxis of VTE following a single administration.
A phase I randomized, double-blind, placebo-controlled, single-dose, dose-escalation study evaluated the safety and the PK/PD profile of increasing IV doses of TB-402 (between 0.015 and 1.860 µg/kg) in healthy young and older volunteers [Verhamme et al. 2010]. A plateau effect in terms of FVIII level inhibition was observed. At plateau, FVIII levels were generally decreased by 1/3 to 2/3 from baseline. aPTT prolongation (generally 1.1–1.2 times baseline) was also observed and maintained for at least 4 weeks, whereas the PT was not modified. The study drug was well tolerated.
A phase II, multicentre, dose-escalating, randomized, enoxaparin-controlled open-label trial was conducted to evaluate the efficacy and safety of a single IV administration of TB-402 for the prevention of VTE in 316 patients undergoing TKR surgery [Verhamme et al. 2009/2010]. All patients received enoxaparin 40 mg pre-operatively and were randomized postoperatively in a sequential cohort design to one of three doses of TB-402 (0.3, 0.6 or 1.2 mg/kg) administered as an IV bolus 18–24 h after TKR or OD doses of SC enoxaparin 40 mg for at least 10 days (n = 75 per group). Primary end point (asymptomatic DVT as detected by bilateral venography and symptomatic VTE by day 7–11) was observed in 16.7%, 23.9%, 24.1% and 39.0% patients for TB-402 doses of 0.3 mg/kg, 0.6 mg/kg, 1.2 mg/kg and enoxaparin 40 mg, respectively. Major or clinically relevant nonmajor bleeding (main safety outcome) was observed in 4.0%, 5.4%, 8.0% and 3.8% patients for TB-402 doses of 0.3 mg/kg, 0.6 mg/kg, 1.2 mg/kg and enoxaparin 40 mg, respectively (p = NS). In summary, TB-402 showed no significant dose–response trend in the reduction in rates of VTE at doses between 0.3 and 1.2 mg/kg but there was a dose–response trend for clinically relevant bleeding. The lower TB-402 dose (0.3 mg/kg) showed a good efficacy with no significant increase in clinically relevant bleeding compared with enoxaparin 40 mg. Further phase III confirmatory trials are needed.
Factor VIIa/tissue factor inhibitors
The complex formed between FVIIa and TF (VIIa/TF) plays a crucial role in the initiation of the coagulation cascade in response to an injury at the vessel wall. New parenteral drugs targeting VIIa/TF (inhibiting the initiation of coagulation) include tifacogin (recombinant tissue factor pathway inhibitor [TFPI]), NAPc2 (recombinant nematode anticoagulant peptide), PCI-27483 and BMS-593214.
Tifacogin (rTFPI, TFP561)
Tifacogin (Novartis) is a recombinant form of human TFPI. It is produced in Escherichia coli and is distinguished from endogenous TFPI by an alanine at the N terminus and lack of glycosylation [Gustafson et al. 1994]. TFPI may protect the microvasculature endothelium from coagulation and sepsis-induced injury, as supported by several preclinical studies [Carr et al. 1995; Camerota et al. 1998]. Tifacogin was found to be well tolerated with no clinically significant bleeding in healthy volunteers when administered as an infusion in doses of 0.5–1.8 mg/kg/h for 72 h [Creasey, 1999].
In a phase II trial, 210 sepsis patients were randomized to receive one of two doses of tifacogin (25 or 50 µg/kg/h) by continuous infusion or placebo for 4 days. Mean rTFPI levels during infusion were proportional to the dose. TFPI concentrations returned to predose values by 4 h after the end of infusion. Compared with placebo, tifacogin produced a 20% relative reduction in 28-day mortality. Major bleeding occurred in 9% and 6% of patients treated with tifacogin and placebo, respectively [Abraham et al. 2001].
A phase III trial called OPTIMIST (Optimized Phase III Tifacogin in Multicenter International Sepsis Trial) compared tifacogin with placebo in 1754 severe sepsis patients [Abraham et al. 2003]. The primary end point, 28-day mortality, was similar with tifacogin (34.2%) and placebo (33.9%), while bleeding rates were higher with tifacogin (6.5%) than with placebo (4.8%). A retrospective analysis of 847 patients with sepsis due to community-acquired pneumonia (CAP) from the OPTIMIST trial showed an improved survival in patients who did not receive concomitant heparin (29.3% vs. 51.9%) [Laterre et al. 2009]. A phase III clinical trial (CAPTIVATE: ‘Community Acquired Pneumonia Tifacogin Intra Venous Administration Trial for Efficacy’) comparing two doses of tifacogin with placebo in 2136 patients with severe CAP was completed as for July 2008 [ClinicalTrials.gov, NCT00084071]. Protocol details have been published [Laterre, 2008] but the results have not yet been reported.
rNAPc2 (recombinant nematode anticoagulant peptide)
rNAPc2 (ARCA Biopharma) is a recombinant nematode anticoagulant protein c2 originally isolated from the canine hookworm, Ancylostoma caninum [Cappello et al. 1995]. rNAPc2 has a half-life of approximately 50 h after SC injection [Vlasuk et al. 2003]. Initial clinical trials with NAPc2 focused on VTE thromboprophylaxis [Lee et al. 2001]. Further studies focused NAPc2 on ACS [Moons et al. 2003; Giugliano et al. 2007]. An additional phase I–II safety study of rNAPC2 to prevent tumour progression and metastases in metastatic colon cancer was started in December 2006 but the study has been terminated because the laboratory has recently suspended the development of the product [ClinicalTrials.gov, NCT00443573].
PCI-27483
PCI-27483 (Pharmacyclics) is a small molecule of undisclosed structure with high potency and selectivity for the FVIIa/TF complex. Preclinical studies showed the antitumour effects of PCI-27483 [Prescott et al. 2008; Holsinger et al. 2006]. The antithrombotic effects of PCI-27483 were determined in a baboon model of arterial thrombosis. PCI-27483 showed dose-dependent inhibition of thrombus formation, fibrin accumulation and PT. PCI-27483 4 mg/kg showed comparable anticoagulation effects as 2 mg/kg enoxaparin [Pharmacyclics, 2009].
A phase I study in healthy volunteers was conducted to assess the PD and PK profiles of PCI-27483 following a single, SC injection. The half-life of PCI-27483 was 10-12 h. The International Normalized Ratio (INR) was strongly correlated with drug plasma concentration. PCI-27483 was well tolerated [Pharmacyclics, 2009]. This compound is being evaluated in a phase Ib/II trial in patients with pancreatic cancer receiving treatment with gemcitabine [ClinicalTrials.gov, NCT01020006].
BMS-593214
BMS-593214 (Bristol Myers Squibb) is a synthetic, competitive, direct and potent inhibitor of FVIIa with a Ki of 0.4 nM [Wong et al. 2010]. The enzyme kinetics, antithrombotic and antihaemostatic properties of BMS-593214, an active-site, direct FVIIa inhibitor were evaluated in enzymatic assays and in anaesthetized rabbit models of electrically-induced carotid arterial thrombosis, thread-induced vena cava venous thrombosis and cuticle bleeding time (BT). BMS-593214 showed selectivity for FVIIa and exhibited species differences in TF-FVIIa-dependent anticoagulation with similar potency in human and rabbit plasma. BMS-593214 was efficacious in the prevention and treatment models of arterial thrombosis and venous thrombosis with ED50 values of 1.1 to 3.1 mg/kg. BMS-593214 exhibited a wide therapeutic window with respect to BT.
Factor Va inhibitors
FVa is a key cofactor in thrombin generation. It acts as a cofactor of FXa (both form the prothrombinase complex) that, in the presence of Ca++, convert prothrombin to thrombin on the cell surface membrane. FVa is inhibited by activated protein C. FVa inhibitors include drotrecogin alpha activated (DrotAA) and ART-123. These drugs have been mainly developed to improve the management of patients with severe sepsis and/or disseminated intravascular coagulation (DIC), but ART-123 has also been tested for thromboprophylaxis after THR.
Drotrecogin alpha activated (recombinant activated protein C)
DrotAA (Xigris®, Eli Lilly) is a recombinant form of activated protein C licensed for treatment of adult patients with severe sepsis with multiple organ failure when added to best standard care [European Medicines Agency, 2010e]. Animal models showed that the administration of activated protein C may improve the outcome of severe sepsis [Taylor et al. 1987]. Half-life is approximately 13 min in adults and 30 min in children [Barton et al. 2004]. The plasma clearance of DrotAA is approximately 41.8 l/h in sepsis patients as compared with 28.1 l/h in healthy subjects [European Medicines Agency, 2010e].
In a placebo-controlled phase II trial in patients with severe sepsis, an infusion of DrotAA resulted in dose-dependent reductions in the plasma levels of D-dimer and serum levels of interleukin-6, markers of coagulopathy and inflammation, respectively [Hartman et al. 1998]. The approval of the indication in adult patients with severe sepsis was based on the results of the PROWESS trial (Recombinant Human Activated Protein C Worldwide Evaluation in Severe Sepsis), involving 1690 subjects [Bernard et al. 2001]. DrotAA (24 µg/kg/h IV infusion for 96 h) showed a 19.4% reduction in the relative risk of death from all causes and an absolute reduction of 6.1% at 28 days as compared with placebo, while the incidence of serious bleeding was higher in the DrotAA group than in the placebo group (3.5% vs. 2.0%; p = 0.06). An additional phase III study (ADDRESS study: ‘Administration of Drotrecogin Alfa (Activated) in Early Stage Severe Sepsis’) was terminated early due to increased bleeding risk and no benefit in mortality in adults with sepsis at low risk of death [Abraham et al. 2005]. The international PROGRESS registry involved outcome data from patients with severe sepsis [Martin et al. 2009]. Patients receiving DrotAA had a 28% (95% CI, 0.60–0.86) reduction in the odds of death and a RR reduction of 17% (p = 0.0003). In an additional prospective, observational study of Xigris (DrotAA) use in the United States (XEUS study), a total of 548 adult patients with severe sepsis were enrolled [Steingrub et al. 2010]. Patients receiving DrotAA in clinical practice were more acutely ill, had a higher mortality rate, but a similar safety profile compared with the PROWESS trial.
Data from studies in children with sepsis indicates a degree of coagulopathy and DrotAA PK similar to that observed in adults, with bleeding the main safety concern [Nadel et al. 2007; Goldstein et al. 2006; Barton et al. 2004].
ART-123 (recombinant thrombomodulin alpha)
ART-123 (Recomodulin®; Artisan Pharma, licensed from Asahi Kasei Pharma Corporation) is a recombinant human soluble thrombomodulin. ART-123 binds to thrombin to inactivate coagulation, and the thrombin-ART-123 complex activates protein C. ART-123 showed a favourable antithrombotic profile with less bleeding than other anticoagulants in animal and in vitro experiments [Mohri et al. 1999, 1997]. In healthy volunteers, the plasma half-life ranged between 20 h [Nakashima et al. 1998] and 2–3 days [Moll et al. 2004]. Antithrombotic activity is maintained for at least 6 days after administration of ART-123 [Moll et al. 2004].
A phase II open-label, dose-ranging study was performed in which 312 patients undergoing THR received either 0.3 mg/kg (low-dose) or 0.45 mg/kg (high-dose) of ART-123, SC, 2-4 h after surgery (day 1) [Kearon et al. 2005]. The primary efficacy outcome (all DVT on mandatory bilateral venography performed on day 9 ± 2 and symptomatic VTE up to day 11) occurred in 3.4% of 116 evaluable patients in the ART-123 low-dose group and 0.9% of 111 patients in the ART-123 high-dose group. Major bleeding occurred in 1.4% and 6.3% of patients in the ART-123 low- and high-dose groups, respectively. ART-123 has not been further developed for thromboprophylaxis.
In a phase II clinical assessment of ART-123 in sepsis, the compound showed a good dose–response trend in patients with DIC [Maruyama, 1999]. A confirmatory phase III multicentre, double-blind, randomized, parallel-group trial compared the efficacy and safety of IV infusions of ART-123 (0.06 mg/kg for 30 min, OD) to those of low-dose heparin (8 IU/kg/h for 24 h) for 6 days in 234 of DIC patients [Saito et al. 2007]. The primary efficacy end point (DIC resolution rate) occurred in 66.1% of the ART-123 group as compared with 49.9% of the heparin group [difference, 16.2%; 95% CI, 3.3–29.1%]. Rates of bleeding-related adverse events were lower in the ART-123 group than in the heparin group (43.1% vs. 56.5%, p = 0.0487). On the basis of these results, ART-123 (Recomodulin®) has been licensed for the treatment of DIC in Japan as for January 2008 (Artisanpharma press release, 22 February 2008, www.artisanpharma.net). There is an ongoing randomized, double-blind, placebo-controlled, phase IIb study to assess the safety and efficacy of ART-123 on subjects with sepsis and DIC that will enrol 800 subjects in over 140 sites worldwide [ClinicalTrials.gov, NCT00487656].
Dual FXa/thrombin inhibitors
A dual-acting Xa/thrombin inhibitor might provide a better overall profile than either mechanism alone by the prevention of thrombin formation through the anti-FXa activity and simultaneous inhibition of thrombin activity.
EP217609
EP217609 (Endotis Pharma, licensed from Organon/Schering Plough) is a new synthetic parenteral dual-action anticoagulant combining an indirect FXa inhibitor (antithrombin binding pentasaccharide), a direct thrombin active site inhibitor (peptidomimetic, α-NAPAP derivative) and biotin, which allows its neutralisation by avidin, an egg-derived glycoprotein [Petitou et al. 2009]. Anti-Xa IC50 is 11.50 nM and anti-IIa IC50 is 11.48 nM. The half-life in rats is 2.9 h and SC bioavailability is 100%. In experimental venous and arterial thrombosis models, the drug was more potent than UFH, fondaparinux and argatroban on a molar basis and had an enhancing effect on surgically induced bleeding [Petitou et al. 2009]. Avidin triggered a rapid, complete and irreversible neutralization of EP217609 without rebound effect in a canine model of extracorporeal circulation [Fromes et al. 2010].
EP217609 was well tolerated in 24 healthy subjects exposed to single, ascending IV boluses of 1, 3 and 10 mg. There was a dose-dependent increase in exposure and also in ACT, TT, ECT, PT, aPTT and thrombin generation test (TGT) lag time with a low intersubject variability. Maximum anticoagulant effect was reached within 5 minutes after bolus injection and lasted for up to 3 days. EP217609 was partially eliminated by the kidney as unchanged drug [Gueret et al. 2009/2010]. In a separate trial, avidin was well tolerated in 24 healthy subjects exposed to single, ascending doses. There was no evidence of hypersensitivity reactions. Phase II trials are planned in two target indications: extracorporeal circulation in cardiac surgery and PCI in ACS [Endotis press release, 4 June 2010, www.endotis.com].
Tanogitran (BIBT 986)
Tanogitran (Boehringer Ingelheim), a novel synthetic, small-molecule (MW 542 Da) is a specific, direct, competitive and reversible inhibitor of both, FXa (Ki 26 nM) and thrombin (Ki = 2.7 nM). BIBT 986 showed a high anticoagulant potency in preclinical models of venous thrombosis in rats and rabbits [Graefe-Mody et al. 2006].
In three randomized, double-blind, placebo-controlled trials, subjects were administered by IV infusion escalating doses of BIBT 986 (from 0.2 to 5 mg/kg) for up to 32 h [Graefe-Mody et al. 2006]. In all three studies, IV infusion of BIBT 986 was safe and well tolerated. BIBT 986 exhibited linear PK over the dose range tested. Clearance was about 8 l/h and VD about 50 l. Apparent steady-state concentrations were reached within 24 h, indicating a dominant half-life of about 6 h. The terminal half-life of BIBT 986 was approximately 12 h. Renal excretion contributed approximately 50% to total elimination. Overall interindividual variability in PK and PD parameters was <40%. There was a linear correlation between plasma concentrations and PD responses.
In a prospective, randomized, double-blind, placebo-controlled, parallel-group, dose-escalation trial in 48 healthy male volunteers, the effect of three IV doses of BIBT986 on coagulation, platelet activation, and inflammation was compared with that of placebo IV together in a human endotoxemia model with a bolus infusion of 2 ng/kg lipopolysaccharide (LPS) [Leitner et al. 2007]. BIBT986 doses, which prolonged aPTT by 100%, completely suppressed the LPS-induced increases in prothrombin fragment, thrombin–antithrombin complexes, and D-dimer as compared with placebo. BIBT986 did not influence inflammation, fibrinolysis, or platelet activation.
Potential benefits of new parenteral anticoagulants
Potential benefits and drawbacks of parenteral anticoagulants in development versus UFH.

UFH, unfractionated heparin; PK/PD, pharmacokinetics/pharmacodynamics; UNK, unknown.
Bleeding is the main adverse effect of anticoagulation. The availability of an antidote for RB006 (RB007), idrabiotaparinux and EP217609 (avidin), M118 (protamine), and the rapid offset of action of otamixaban (Table 3) are positive features in situations in which coagulation needs to be rapidly restored (i.e. after an interventional procedure or if bleeding occurs during anticoagulant administration).
Most of the new anticoagulants are synthetic (i.e. otamixaban, idrabiotaparinux, anticoagulant aptamers), thus minimizing the risk for contamination during manufacturing process compared with animal-derived substances [Alban, 2005].
HIT is a serious complication of heparin therapy [Warkentin et al. 2008], which is usually caused by the production of antibodies to a complex of heparin and platelet factor-4 (PF4). Alternatives with a grade 1 recommendation for treatment of HIT are parenteral danaparoid, lepirudin and argatroban [Warkentin et al. 2008]. New parenteral synthetic anticoagulants, such otamixaban, RB006 or new parenteral thrombin inhibitors may be potentially useful for anticoagulation in patients with history of HIT. The potential for HIT with new substances based on the pentasaccharide sequence present in heparin seems to be low for semuloparin and M118, and very low or negligible for synthetic idrabiotaparinux and EP217609.
ULMWH retains LMWH properties with respect to inhibition of tumour growth by regulating angiogenesis and apoptosis [Hoppensteadt et al. 2008; Vignoli et al. 2007]. These characteristics may be of interest in the management of cancer-associated thrombosis and prolongation of survival. Two ongoing studies are assessing the efficacy and safety of thromboprophylaxis with semuloparin in cancer patients undergoing major abdominal surgery (SAVE-ABDO, 4400 patients) [ClinicalTrials.gov, NCT00679588] or undergoing chemotherapy (SAVE-ONCO, 3200 patients) [ClinicalTrials.gov, NCT00694382] (Table 2).
The limited renal excretion of otamixaban and RB006 compared with conventional anticoagulants that are mainly cleared through the kidneys (i.e. LMWH, fondaparinux, hirudins) may represent an advantage in patients with renal insufficiency. Specific studies with otamixaban are currently ongoing in patients with severe renal impairment [ClinicalTrials.gov, NCT01120314] and in patients with mild and moderate hepatic impairment [ClinicalTrials.gov, NCT01126086].
Potential drawbacks of new parenteral anticoagulants
Given that most of the new anticoagulants are in a preclinical or early clinical phase of development, there is limited information on the safety and efficacy compared with conventional anticoagulants. In particular, the information is very limited for potential drug–drug interactions and special populations, such as patients with renal or hepatic insufficiency, children or pregnant women. Current anticoagulants (i.e. UFH, LMWH or fondaparinux) accumulate in renal insufficiency. Owing to their renal elimination, idrabiotaparinux, EP217609, ULMWH and M110 are also likely to accumulate in renal insufficiency.
The lack of specific antidote is a drawback of some of the newer parenteral anticoagulants in development (i.e. otamixaban, direct thrombin inhibitors). Laboratory investigations suggest that recombinant factor VIIa (rFVIIa) may be effective in reversing the effects of anticoagulant therapy [Vavra et al. 2010]. However, owing to the limited efficacy data with rFVIIa in this setting [Vavra et al. 2010] and the risk of thrombosis [Gill et al. 2009; Mayer et al. 2008], further studies are required before specific recommendations could be made.
Rebound anticoagulation has been described after dissociation of the heparin–protamine complex (rebound heparin activity), limiting durability and the desired clinical effect of protamine in some cases [Teoh et al. 2004]. Avidin (the antidote of idrabiotaparinux) has a short half-life (minutes) versus idrabiotaparinux (weeks). Idrabiotaparinux may return into the circulation from its third distribution departments of tissues into blood after several weeks resulting in rebound anticoagulant effect [Harenberg, 2009].
Rebound hypercoagulation (increase in thromboembolic events after anticoagulants are stopped) has been described in the literature [Hermans and Claeys, 2006; Ansell, 2007]. Further clinical data with new parenteral anticoagulants are needed on this regard.
Some of the new anticoagulants (i.e. semuloparin and M118) are obtained by depolymerization of UFH heparin from pig intestinal mucosa. The heparin contamination crisis (certain batches of heparin that were associated with an acute, rapid onset of serious side effects indicative of allergic-type reactions due to the presence of the contaminant oversulfated chondroitin sulfate [OSCS], which was not removed by any of the processes used to make LMWH from heparin), has highlighted the importance of designing and implementing appropriate analytical tests as controls, tests which are able to measure molecular properties of the complex mixture [Guerrini et al. 2009; Sasisekharan and Shriver, 2009; Zhang et al. 2008].
Hypersensitivity reactions are off-target effects reported during heparin [Hirsh et al. 2008] or protamine administration [Nybo and Madsen, 2008]. Off-target effects cannot also be ruled out with new anticoagulants or their antidotes. Anticoagulant oligonucleotides (i.e. aptamers such as RB006, NU172 or HD1-22, and antisense oligonucleotides) may potentially induce pyrexia and haemodynamic instability following IV administration (largely mediated by the alternative complement pathway) and innate immune stimulation [Keefe et al. 2010]. In addition, RB006 is conjugated to PEG. Antibodies to synthetic oligonucleotides are not generally observed, but have been seen for some oligonucleotide conjugation partners such as PEG [Armstrong et al. 2007]. For therapeutic proteins (i.e. DrotAA, ART-123, tifacogin, rNAPC2) or monoclonal antibodies (i.e. TB-402), as with all therapeutic proteins, there is a potential for immunogenicity. However, the risk for immunogenicity with DrotAA, as measured by the presence of antibodies after treatment, seems to be low [Yan et al. 2009]. On the other hand, the use of antidotes such as avidin (an egg-derived protein) or RB007 (an oligonucleotide) may not be free from risk of development of antibodies and/or hypersensitivity reactions [Keefe et al. 2010; Paty et al. 2010].
Despite the fact that new anticoagulants have been developed to be given without routine anticoagulant monitoring, it may still be helpful in some circumstances, such as pregnancy or renal impairment [Ansell et al. 2008; Hirsh et al. 2008; Michota and Merli, 2005]. Idrabiotaparinux and ULMWH can be monitored using modified anti-FXa assays, but the therapeutic range is pending to be established. Like conventional heparins, M118 anticoagulant activity can be monitored using a chromogenic anti-FXa activity assay.
The cost of the new parenteral anticoagulants in development is likely to be higher than that of UFH and other current therapeutic options. In particular, for aptamers and antisense oligonucleotides, their high molecular mass and complex syntheses make them more expensive to manufacture than small molecules [Keefe et al. 2010].
Parenteral anticoagulants in development versus new oral anticoagulants
As discussed previously, the new parenteral anticoagulants in development are expected to further refine current parenteral anticoagulation. Parenteral anticoagulants will still play an important role in therapeutics when a rapid onset and offset of anticoagulation is needed (i.e. ACS, anticoagulation during extracorporeal circulation, prevention of clotting during haemodialysis). New oral anticoagulants (i.e. dabigatran, rivaroxaban, apixaban, among others) are being mainly developed to replace warfarin for long-term anticoagulation (i.e. stroke prevention in AF, long-term treatment of VTE) without the need for routine anticoagulant monitoring [Ahrens et al. 2010; Eikelboom and Weitz, 2010; Gómez-Outes et al. 2009]. Compared with new oral anticoagulants, new parenteral anticoagulants do not appear able to bring significant benefit for long-term anticoagulation, as shown by the discontinuation of the clinical development of idrabiotaparinux in patients with AF (Sanofi-Aventis press release, 21 December 2009, www.sanofi-aventis.com).
There are a number of indications currently dominated by parenteral LMWH and fondaparinux (i.e. VTE prophylaxis and treatment) in which a gradual and partial replacement by new oral anticoagulants is expected. An oral medication may simplify outpatient thromboprophylaxis and treatment, and may be preferred by patients compared with daily SC injections, but education and the detection of risk factors of nonadherence to treatment are still essential [Bellamy et al. 2009]. Oral dabigatran and rivaroxaban are licensed for thromboprophylaxis in THR and TKR [European Medicines Agency, 2010b, 2010d]. New oral anticoagulants are not currently available for thromboprophylaxis in medical patients, although confirmatory trials in these patients are ongoing with rivaroxaban and apixaban [Gómez-Outes et al. 2009]. New oral anticoagulants have not been tested for thromboprophylaxis in HFS or major abdominal surgery. In these patients, LMWH and fondaparinux will remain the treatment of choice in the near future. Semuloparin, a new ULMWH, has the potential to be an alternative to LMWH and fondaparinux in VTE prophylaxis in THR, TKR, HFS and in patients with cancer, but additional results from ongoing studies are required. Rivaroxaban and apixaban are being tested for initial VTE treatment without overlapping with a parenteral anticoagulant during the first days [Gómez-Outes et al. 2009], but dabigatran will need overlapping with a parenteral anticoagulant for the first few days after acute VTE [Schulman et al. 2009] if the approval for this indication is granted. The results of the ongoing CASSIOPEA study in patients with acute PE [ClinicalTrials.gov, NCT00345618] will be necessary to ascertain whether once-weekly SC idrabiotaparinux may be an alternative to established treatment in patients with acute VTE. Finally, it is unlikely that new oral anticoagulants (i.e. dabigatran and rivaroxaban) would replace heparins for anticoagulation during pregnancy [Bates et al. 2008] because reproductive toxicity has been shown in animal studies with these new compounds [European Medicines Agency, 2010b, 2010d]. Further data on the use of new parenteral anticoagulants during pregnancy are also required.
Conclusions
New parenteral anticoagulants have the potential to complement established anticoagulants for anticoagulation in VTE (i.e. semuloparin, idrabiotaparinux), ACS (i.e. otamixaban, M118) and/or may represent an alternative to the anticoagulant/antidote pair heparin/protamine during cardiac surgery (i.e. RB006/RB007, EP217609/avidin). Results of confirmatory trials with these new parenteral anticoagulants are needed before establishing their place in therapeutics.
Footnotes
Acknowledgement
The views and opinions expressed in this review are of the authors alone and do not necessarily represent the official views of their institutions or any other party.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors declare no conflicts of interest.