
Abstract
Spontaneous pneumothorax (SP) is defined by the presence of air in the pleural space arising from neither trauma nor iatrogenic causes. It can develop secondary to various etiologies. The familial clustering observed in some patients with SP supports the view that genetic factors play a role in the pathogenesis of SP. Several recognizable pneumothorax-associated genetic syndromes exist, including Birt-Hogg-Dubé syndrome (BHD), tuberous sclerosis complex-associated lymphangioleiomyomatosis (TSC-LAM), Marfan syndrome (MFS), cystic fibrosis (CF), alpha-1 antitrypsin deficiency (AATD), vascular Ehlers-Danlos syndrome (vEDS), Loeys-Dietz syndrome (LDS), and a few others. Recognition of these syndromes underlying SP facilitates optimal management and counseling regarding prognosis and potential comorbidities. In this review, we systematically summarize several genetic syndromes associated with pneumothorax, in which SP may manifest either as an initial presenting symptom or as a potential complication that adversely affects the prognosis.
Keywords
Introduction
Pneumothorax, defined as the presence of air in the pleural cavity, is a common clinical problem worldwide. It can develop secondary to various etiologies, including traumatic, iatrogenic, inflammatory, infectious, malignant, genetic, and hormonal causes. Spontaneous pneumothorax (SP) is characterized by air in the pleural space without trauma or iatrogenic cause. SP remains a significant health problem, and the incidence varies considerably. Bense et al. found an incidence of SP in Sweden of 18–28 per 100,000 in men and 1.2–6 in women. 1 A retrospective, descriptive epidemiological study based on a national healthcare database in England showed that the overall incidence of SP was 16.8 cases per 100,000 population per year (24/100,000/year for males; 9.8/100,000/year for females). 2 In a recent study in France, the annual incidence of SP was estimated at 22.7 cases per 100,000 inhabitants. 3 It is worth noting that the prevalence of SP is not fully known, as some patients may be asymptomatic, but the incidence has been shown to be increasing. 4 Current understanding regarding the epidemiology of SP has been informed by small studies performed at single medical centers or retrospective reviews of national data registries. Overall, the incidence of SP has been estimated to be 17–24/100,000 in the male population and 1–6/100,000 in the female population. 5
While the majority of SP are sporadic and attributable to environmental or acquired factors, emerging evidence indicates that a subset of cases cluster within families and is associated with identifiable genetic or syndromic conditions. 6 Familial spontaneous pneumothorax (FSP) accounts for 10% of primary SP. 7 Several recognizable pneumothorax-associated genetic syndromes exist, such as Birt-Hogg-Dubé syndrome (BHD), tuberous sclerosis complex-associated lymphangioleiomyomatosis (TSC-LAM), Marfan syndrome (MFS), cystic fibrosis (CF), alpha-1 antitrypsin deficiency (AATD), vascular Ehlers-Danlos syndrome (vEDS), Loeys-Dietz syndrome (LDS), homocystinuria, cutis laxa, Sotos syndrome, among others. 8 Proper diagnosis of these syndromes facilitates optimal management and counseling regarding prognosis and potential comorbidities. In this review, we systematically summarize several genetic syndromes associated with pneumothorax, in which SP may manifest either as an initial presenting symptom or as a potential complication that adversely affects the prognosis. 8
Birt-Hogg-Dubé syndrome
BHD is a rare inherited autosomal dominant disorder caused by germline mutations in the tumor suppressor gene FLCN, which encodes the protein folliculin. 9 Its clinical expression typically includes multiple pulmonary cysts, recurrent SP, cutaneous fibrofolliculomas, and renal tumors of various histological types. Patients with BHD typically exhibit multiple bilateral pulmonary cysts (Figure 1(a)), multifocal and/or bilateral renal tumors, often with diverse and occasionally mixed histopathological features (Figure 1(b)), and skin lesions that commonly present as small, soft, skin-colored benign tumors, predominantly distributed on the eyelids, neck, and axillae (Figure 1(c)).

Clinical manifestations of BHD. (a) Chest CT reveals bilateral cystic lesions with thin walls and varying sizes, predominantly distributed in the lower lobes, particularly adjacent to the mediastinum and subpleural regions. (b) Contrast-enhanced renal CT demonstrates a well-defined, homogeneous, round soft tissue mass in the mid pole of the right kidney (red arrow), with pathology confirming clear cell carcinoma. (c) Multiple skin-colored papules on the neck (red arrows), with pathology confirming fibrofolliculomas.
Grimes et al. performed systematic genetic testing in 72 patients with FSP, achieving etiologic diagnoses in 19 of cases (26.4%). BHD was the most prevalent etiology, constituting 47% of the genetically confirmed diagnoses. 7 A retrospective study conducted by the thoracic surgeon team at Peking University People’s Hospital revealed that almost 2/3 of hereditary pneumothorax were diagnosed with BHD following Sanger genetic testing. 10 Yngvadottir et al. demonstrated that the frequency of clinically validated loss-of-function FLCN variants is 1 in 2710–4190. The lifetime risk of pneumothorax in FLCN mutation carriers was substantial (28.4%–37.3% to age 65 years). 11 Studies from both China and the Netherlands have shown BHD to account for 5%–10% of patients presenting with an apparent SP. 12
Over 80% of patients with BHD eventually manifest multiple bilateral pulmonary cysts on chest computed tomography (CT), mainly located at the base of the lungs, often adjacent to the pleura or mediastinum, with normal surrounding parenchymal tissue (Figure 1(a)).9,13 The mechanisms leading to the formation of pulmonary cysts following loss of folliculin function are not well defined. A recent hypothesis suggests that cysts could arise as a consequence of deficient cell-cell adhesion and repeated stretch-induced stress caused by breathing, thus leading, over the long term, to the expansion of alveolar spaces, predominantly in lung regions where alveolar volume changes during the respiratory cycle are more pronounced. 14 This “stretch hypothesis” potentially provides an explanation for the predominantly subpleural and basal distribution of pulmonary cysts in BHD.
Due to the propensity of cysts to rupture, affected individuals are predisposed to SP with a high recurrence rate. There is an estimated 50-fold higher risk of SP in patients with BHD compared to the general population. 15 The mean age of occurrence for the first episode of pneumothorax in patients with BHD was between 36 and 38 years in three large cohorts.13,16,17 The recurrence rate was 75%–82% with an average number of 3.6 episodes.13,16,17 The presence, number, size and total volume of pulmonary cysts, along with a family history of pneumothorax, have been identified as risk factors for the occurrence of pneumothorax in patients with BHD. 18 The previous study by Hu et al. has shown that the most common manifestations of BHD in China were multiple pulmonary cysts (92.4%), SP (71.0%), skin lesions (18.1%), and renal tumors (3.6%). 19 A family history of SP was identified in 84.7% of the BHD families, with an average number of SP being 1.8 (range: 1–6). 19 Pulmonary cysts and/or pneumothorax have no sex predilection and are not associated with smoking or the presence and/or severity of cutaneous and renal involvement.13,15 Another study by our team demonstrated that the long-axis diameter, short-axis diameter, and volume of the largest cysts were associated with the occurrence of SP. 20
In recent years, studies have further analyzed the correlation between FLCN gene variants and the occurrence of pneumothorax. A German cohort study showed that FLCN pathogenic variants c.250-2A>G (77%) and c.1300G>C (59%) significantly increased the risk of pneumothorax in BHD compared with variant c.1285dupC (37%). 21 The latest analysis by Hu et al. on the correlation between genes and phenotypes in 76 cases of Chinese patients with BHD, found a significant difference in the incidence of pneumothorax between exon 1–3 large segment deletions and point variants, with rates of 91% and 58%, respectively, with a p-value of 0.047. 22 Patients with BHD also have an increased risk of pneumothorax while diving, as well as during and following air travel, with a flight-related pneumothorax rate ranging from 0.12% to 0.63% per flight.16,23,24 This could be explained by changes in atmospheric pressure in the aircraft cabin during flights, which can cause cyst expansion and predispose to rupture.
Due to the high recurrence rate of SP, pleurodesis is recommended with the first episode of SP. 25 Liu et al. demonstrated that the risk of recurrence of SP in BHD after surgical treatment was 9.1%, significantly lower than the 53.1% recurrence rate with conservative management. 10 A study from the United States showed similar results, with pleurodesis reducing the risk of recurrent ipsilateral pneumothorax by approximately 50%. 16 A recent retrospective study by Seyama et al. which included 81 cases of pneumothorax complicated by BHD, demonstrated that a total pleural covering of the entire visceral pleura with oxidized regenerated cellulose mesh significantly reduced the recurrence rate of pneumothorax in patients with BHD, with the 5-year recurrence rate of 12% after partial pleural covering, and no pneumothorax recurrence with total pleural fixation at a median follow-up of 34 months in (p = 0.032). 26
Tuberous sclerosis complex-associated lymphangioleiomyomatosis
Lymphangioleiomyomatosis (LAM) is a rare, slowly progressive neoplastic cystic lung disease. It is a mesenchymal tumor composed of histologically and immunohistochemically distinctive perivascular epithelioid cells (PECs), called LAM cells. LAM occurring in patients with underlying tuberous sclerosis complex (TSC) is termed TSC-LAM. LAM can also occur sporadically in women who do not have TSC, known as sporadic LAM. 27 LAM in both forms typically presents in women between the third and fourth decades of life, with a mean age at onset of 34 years. 28 However, it can be encountered in women of any age, ranging from young patients to those in their 80s. 29 The incidence rate varies among populations, ranging from 0.23 to 0.31 women per million per year, and is increasing due to advances in disease recognition. 27
TSC-LAM is an autosomal dominant inherited disorder, resulting from mutations in one of two genes, TSC1 (encoding hamartin) or TSC2 (encoding tuberin). The disease is progressive and results from the proliferation of LAM cells leading to parenchymal destruction and the formation of cysts in the lung, lymphangiomyomas (also known as lymphangioleiomyomas, angioleiomyomas, and lymphangiomas) in the lymphatic system, angiomyolipomas (AMLs) in the kidneys and other organs, and chylous effusions in the pleural space and peritoneum.30,31 Multiorgan features occur in TSC affecting the brain, skin, abdominal viscera, heart, eyes, mouth, as well as the lungs. Of adult women with TSC, 26% to 45.5% develop cystic pulmonary lesions identical to those seen in patients with sporadic LAM. Recent studies suggest that lung involvement increase with age, with up to 80% of TSC females affected by age 40.32,33
Patients with TSC-LAM usually show multiple thin-walled, round, well-defined air-filled cysts with preserved or increased lung volume on chest high-resolution computed tomography (HRCT) (Figure 2(a)), and physical examination may reveal accompanying angiofibromas on the face, retroauricular area, and neck (Figure 2(b)–(d)), and/or periungual fibromas (Figure 2(e)). There is no other significant pulmonary involvement, specifically no interstitial lung disease, except for the possible presence of multifocal micronodular pneumocyte hyperplasia in patients with TSC. 34 Progressive dyspnea on exertion and SP are the most common presenting manifestations of LAM. The study by Almoosa et al. showed that 55.5% patients with LAM had a medical history of pneumothorax (56.9% in sporadic LAM vs 47.1% in TSC-LAM, p = 0.29); among subjects who had experienced at least one pneumothorax, the mean number of pneumothorax episodes was 4.4 ± 0.53. 35 Of those with a history of pneumothorax, 4.8% had experienced an episode related to air travel. 35 Risk factors for pneumothorax in patients with LAM include previous pneumothorax, a rapid rate of decrease in FEV1, and large cystic lesions. 29 A study on the risk of recurrent pneumothorax in patients with LAM showed that the median number of pneumothoraxes was 2, with 63% of patients experiencing at least one recurrent pneumothorax. 36 The risk of pneumothorax was higher in LAM patients with an age of onset ⩽35 years and those with a chest CT grade III (highest severity of cystic lung disease), and significantly lower in postmenopausal patients. 36

Clinical manifestations of TSC-LAM. (a) Chest CT demonstrated diffuse thin-walled cystic lesions, ranging in size from several millimeters to a few centimeters. (b–d) Angiofibromas on the face, retroauricular area, and neck. (e) Periungual fibromas.
Patients with LAM are at a higher risk of developing SP during air travel compared to those with other cystic lung diseases. Two survey studies examined the risk of SP with air travel for patients with LAM and estimated the risk of SP per flight to be 1.1% and 2.2%, respectively. 37 Patients with sporadic LAM and TSC-LAM who have well-preserved lung function do not need to take specific precautions or avoid air travel. However, those with advanced diseases should be evaluated for the need for oxygen during flight to prevent hypoxemia and alternative means of travel, as they are less likely to tolerate pneumothorax. Pregnancy exacerbates respiratory symptoms in some women with LAM and appears to be a risk factor for pneumothorax. 38 However, a lack of prospective studies makes the risk difficult to quantify. Polymorphisms in extracellular matrix proteins (collagen, elastin, and matrix metalloproteinase-1) were not associated with pneumothorax in one study. 39
The mechanistic target of rapamycin complex 1 (mTORC1) inhibitor sirolimus lowers the rate of decline of FEV1 in women with LAM. 40 This intervention is based on the discovery that TSC2 mutations result in mTORC1 hyperactivation. Treatment with sirolimus is usually continued indefinitely since lung function has been proven to decline after discontinuation of sirolimus therapy. The impact of sirolimus on pneumothorax risk is not fully known. However, recent research showed that sirolimus treatment was effective in reducing the risk of pneumothorax recurrence, with an 80% reduction in the 5-year risk of pneumothorax recurrence in patients treated compared to those not treated with sirolimus after an episode of pneumothorax. 36 The 5-year risk of pneumothorax recurrence was significantly lower in the sirolimus group, even after excluding patients who had undergone pleurodesis. Therefore, sirolimus is recommended to reduce the risk of pneumothorax recurrence in patients with LAM complicated by pneumothorax. 36 Future studies should further investigate the impact of sirolimus on the risk of pneumothorax in patients with LAM. Other treatments for LAM include oxygen therapy when appropriate, lung transplantation, and avoiding both smoking and estrogen supplementation (may worsen LAM). Progesterone derivatives have been used therapeutically in the treatment of LAM, but without definitive evidence of benefit. 41 Pleurodesis is recommended after the first pneumothorax because of the high likelihood of recurrence.
Marfan syndrome
MFS, an autosomal dominant disorder, is a polymorphic connective tissue disorder. Few autosomal recessive cases have been reported worldwide. More than 90% of MFS cases are associated with mutations in the FBN1 gene, which encodes a 350-kDa extracellular matrix (ECM) glycoprotein representing the principal constituent of the 10-nm microfibrils involved in the maintenance of elastic fibers and in the anchorage of epithelial cells to the interstitial matrix. 42
MFS is best recognized for its associated cardiovascular abnormalities, but it is now understood that it also affects many other organs, including the ophthalmologic and pulmonary systems. Musculoskeletal symptoms include generalized ligamentous laxity, scoliosis (Figure 3(a)), chest deformity, protrusio acetabuli, hypermobility (Figure 3(c) and (d)), foot deformities (Figure 3(e)), dural ectasia, and low bone mineral density. 43 The diagnosis of MFS requires identifying characteristic clinical features and can be made with or without genetic testing for FBN1 pathogenetic variants. 44 The most frequently quoted prevalence of MFS is 20 per 100,000 individuals; Groth et al. found a prevalence of 6.5 per 100,000 in the Danish population. 45
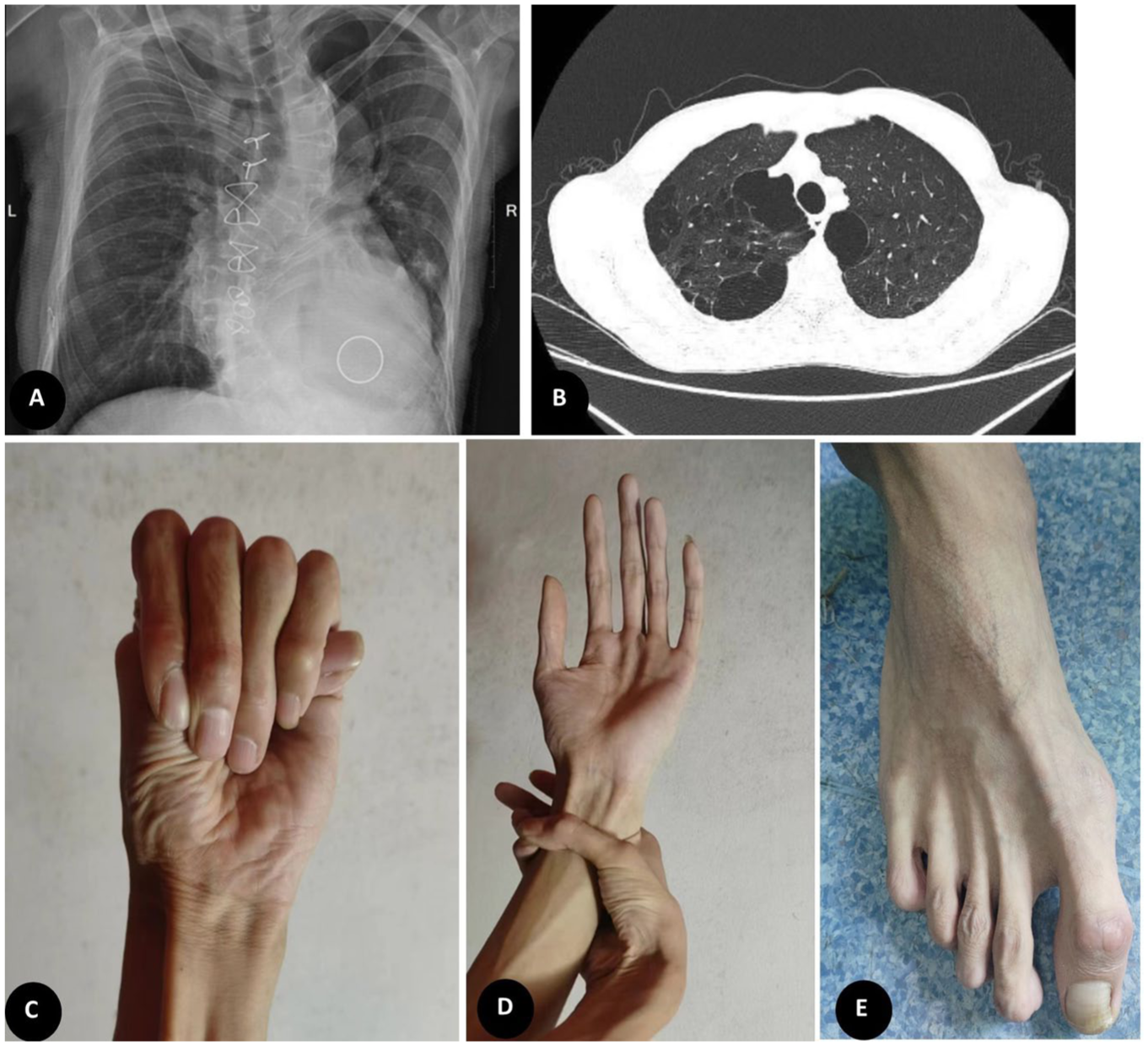
Clinical manifestations of MFS. (a) Chest X-ray revealed significant scoliosis. (b) Chest CT revealed multiple bullae in the lung apices. (c–e) positive thumb and wrist signs and slender toes indicating arachnodactyly.
Mutations in FBN1 gene can induce a variety of effects on the respiratory system, leading to substantial morbidity and potentially increased mortality. These respiratory effects may include chest wall and spinal deformities, emphysema, pneumothorax, sleep apnea, and an increased incidence of asthma, bronchiectasis, and interstitial lung disease. The previous studies published in the 1980s reported the prevalence of pneumothorax in patients with MFS to be 4% and 11%, respectively.46,47 Karpman et al. showed that the frequency of SP was 4.8% in a cohort of patients with a median age of 40 years. 48 Patients with a history of SP were young adults, with a mean age of 21 years at the time of their initial SP, and were more likely to be male. A current or past smoking history did not appear to increase the likelihood of SP.
Grimes et al. also demonstrated that among genetically confirmed cases of FSP, MFS represented the second most frequent etiology (21%), following BHD. 7 The etiology of SP in patients with MFS is controversial. A positive correlation was found between the presence of apical blebs or bullae (Figure 3(b)) and the occurrence of SP, with more than a 9-fold increased risk of pneumothorax in those subjects with identifiable apical blebs or bullae. 48 However, another study showed that although 5%–9.6% of patients with MFS have evidence of blebs on CT, this is not significantly different from the general population. 6 It has been theorized that skeletal deformities may alter the lung parenchymal tension, predisposing individuals to pneumothorax. 49 Karpman et al. reported that not all individuals with MFS have the same increased risk of SP. By using available imaging studies, physicians may be able to stratify patients into higher-and lower-risk groups. 48 If future studies confirm the ability to risk stratify using chest CT scanning, it may be able to de-escalate the concern for SP in many patients with MFS, particularly those without apical blebs or bullae. 48
The recommended management for the first episode of SP in adult patients with MFS is the same as in the general population. 50 However, since the risk of SP is higher in patients with MFS compared to the general population, individuals with MFS are advised to avoid scuba diving, rapid ascents in elevators, and playing brass instruments. 51
Cystic fibrosis
CF is an autosomal recessive genetic condition caused by mutations in the CFTR gene and manifests multiple clinical manifestations, the most prevalent being pancreatic insufficiency with malnutrition and bronchiectasis with chronic airways infection. The natural history of the lung disease consists of early and persistent infection, an exaggerated inflammatory response, and progressive airways obstruction, often resulting in respiratory failure. As airways disease worsens, the likelihood of respiratory complications, such as hemoptysis and pneumothorax, increases. A mutation in CFTR gene changes a protein (a regulated chloride channel), which regulates the activity of chloride and sodium channels at the cell surface epithelium. There are about 70,000 worldwide cases, and approximately 1000 new cases are added each year. CF is most common in white people of northern European ancestry, having 1 in 2000–3000 births, and least in Asian-Americans, affecting 1:30,000 newborns.52,53
A retrospective study over 10 years showed that the average annual incidence of pneumothorax in CF patients was 0.64%–3.4%. 54 For example, Flume et al. showed that the average annual incidence of pneumothorax is 0.64%, or 1 in 167 patients per year. 55 Approximately 3.4% of individuals with CF will experience a pneumothorax during their lifetime. 56 Because patients typically have severe obstructive airways disease, plugging of the distal airways with thick secretions may result in trapping of air within the alveoli. When alveolar pressure exceeds interstitial pressure, air moves into the interstitium, travels along the airways into the hilum (pneumomediastinum), and ruptures into the mediastinal parietal pleura. 55 The incidence of pneumothorax is significantly increased in CF patients with Pseudomonas aeruginosa or Burkholderia cepacian complex, Aspergillus infections, forced expiratory volume in one second of less than 30% of predicted, enteral nutrition, pancreatic insufficiency, allergic bronchopulmonary aspergillosis, and massive hemoptysis. 56
Pleurodesis is generally not recommended for the first episode of pneumothorax in patients with CF, whether large or small. 57 However, patients with a recurrent large pneumothorax should undergo pleurodesis since a large portion of patients (estimated 50–90%) may experience further recurrence. 58 For the patient with CF with a pneumothorax who is undergoing pleurodesis, the preferred method is surgical pleurodesis. It should be noted that some experts have expressed concern about the adverse effects that pleurodesis may have on future candidacy for lung transplantation. 57
Alpha-1-antitrypsin deficiency
Alpha-1-antitrypsin (AAT), the major serine protease inhibitor in the serum, is primarily synthesized by hepatocytes. It is also produced in lower concentrations by neutrophils, mononuclear phagocytes, and epithelial cells in the lung and intestine. 59 AAT deficiency (AATD) is inherited as an autosomal codominant condition caused by pathogenic mutations in SERPINA1 gene. There are currently over 120 identified alleles categorized in a specific coding system, in which inherited AAT variants are classified between “A” and “Z” based on their migration in an isoelectric focusing pH gradient. Approximately 90% of individuals in populations of European descent are homozygous for the normal “wild-type” Pi*M allele (genotype MM) of the SERPINA1 gene which encodes AAT. The predominant deficiency-related alleles are Pi*S and the more severe Pi*Z. These alleles exhibit codominant inheritance, generating diverse genotypes. Plasma AAT levels progressively decline with each deficient allele copy (i.e., MM > MS > MZ > SZ > ZZ). Although homozygous Pi*Z deficiency (ZZ-AATD) is rare, with an estimated prevalence of 1/2500–1/5000, heterozygous genotypes like MZ and SZ occur significantly more frequently, found in up to 1/25 individuals in the general population. 60
The incidence of AATD is 1/2000–5000, similar to or higher than that of CF. 61 AATD primarily involves the lungs, liver, and, less frequently, the skin. Deficiency of the serine protease inhibitor AAT results in decreased antiprotease activity, excessive elastin degradation, and progressive emphysema. In the lung, AATD predisposes individuals to the premature onset of chronic obstructive pulmonary disease (COPD), with 1%–2% of all cases estimated to be due to severe AATD. 62 The premature onset of panacinar emphysema is the most prevalent clinical correlate of AATD and the major cause of morbidity and mortality. SP may be the presenting manifestation of the disease or a complication of known emphysema. Bronchiectasis has also been associated with severe AATD.
Recommendations and guidelines from healthcare organizations, such as the World Health Organization (WHO), the Portuguese Society of Pneumology (SPP), the Spanish Society of Pneumology and Thoracic Surgery (SEPAR), and the American and European Thoracic/Respiratory Societies (ATS/ERS), indicate that all individuals with COPD and adults with nonreversible asthma should be tested for AATD at least once during their lifetime. 63 However, researchers are not unanimous on whether to screen for AATD in patients with SP. Current guidelines do not recommend screening for AATD in patients with SP, although this remains a matter of controversy. A study published by Serapinas et al. revealed 7.7% of studied patients with SP had AAT deficiency phenotypes, including severe deficiency-related ZZ and SZ phenotypes. 64 Daniel et al. reported SP to be observed in patients with an abnormally low level of AAT. 65 Recent study by Dias et al. found that 1.9% SP had severe AATD. 66 The study supported screening for AATD in patients with SP, as the detection rate was at least as high as in other conditions where screening is currently recommended.
Although smoking has long been recognized as an important risk factor for lung disease progression 67 and death 68 in patients with AATD, its effect on pneumothorax risk is unknown. The potential link between smoking and pneumothorax in individuals with AATD might be an important area for future research. Also unknown is whether genotype or augmentation therapy (infusions of AATD) can impact pneumothorax incidence or recurrence. Smoking is a significant risk factor for the progression of lung disease and mortality in AATD. However, its influence on the risk of pneumothorax remains unclear. Similarly, it is uncertain whether genotype or augmentation therapy, involving infusions of A1AT, affects the incidence or recurrence of pneumothorax. 68 Published evidence suggests that patients with AATD do not differ significantly from COPD patients of comparable severity in most clinical aspects. For instance, no current evidence indicates a difference in the risk of pneumothorax between these two populations. Consequently, it is reasonable to infer that the management of pneumothorax in AATD patients should follow the clinical principles established for pneumothorax in COPD. 69
Vascular Ehlers-Danlos syndrome
Ehlers-Danlos syndrome (EDS) corresponds to a heterogeneous group of rare, inherited connective tissue disorders, characterized by joint hyperlaxity, high skin elasticity, and connective tissue fragility. In the general population, the estimated prevalence of EDS is 1/5000. In 2017, the international classification was revised to include 13 subtypes, of which classic EDS, hypermobile EDS, and vascular EDS are the most frequent. With the exception of the hypermobile subtype, a number of disease-causing mutations in genes encoding connective tissue proteins have been identified; this genetic diagnosis has made it possible to identify and differentiate between the various subtypes. 70
vEDS is a rare genetic disease with an autosomal dominant trait, caused by pathogenic variants within the collagen type III alpha-1 chain (COL3A1) gene. The resulting quantitative and qualitative alterations in type III collagen typically lead to organ fragility, expressed clinically in young adults as arterial dissections and ruptures, bowel perforation, gravid uterine, and organ ruptures. 71 The precise incidence and prevalence are not known, and, in part because of its rarity, the diagnosis is often made only after a catastrophic complication or at postmortem examination. Pulmonary complications of vEDS include cavitary lesions, cysts, bullae, fibrous nodules, pneumothorax, hemopneumothorax, and pulmonary hemorrhage. Type III collagen in the lung is expressed in vessels and parenchymal fibroblasts. Thus, pulmonary complications of vEDS are proposed to result from poor tissue integrity. The fibrous nodules, or fibrous pseudotumors, feature osseous metaplasia and may result from inefficient repair after injury to the pulmonary vessels or interstitium, with type I collagen favored over the absent type III collagen.
SP can be an early signal of underlying vEDS, sometimes appearing before intestinal and vascular problems. 72 Pneumothorax or hemopneumothorax is one of the diagnostic criteria for vEDS. In patients with vEDS, the deficiency of type III collagen in pulmonary tissue leads to weakening of the pulmonary parenchyma and subpleural supporting structures, predisposing them to the formation of bullae or subpleural emphysema, which may rupture and result in pneumothorax. 71 Shalhub et al. described a multi-institutional experience in the diagnosis of vEDS from 2000 to 2015. 73 They found that the prevalence of SP/hemothorax in patients with vEDS (including pathogenic COL3A1 variants cohort and clinical diagnosis only cohort) to be 8.7%. 73 Boussouar et al. described a large cohort of vEDS patients with long-term follow-up with CT imaging of disease-related pulmonary involvement. In their study, 12.5% of patients with vEDS had a history of SP. 74 The most common abnormal presentation on lung CT scan was emphysema (32.3%), followed by linear opacities (20.5%), clusters of calcified nodules (6.6%), and cavitary nodules (2.9%), comparable for smokers and non-smokers. Types of COL3A1 variants have been shown to significantly influence the onset of arterial and digestive complications, as well as overall mortality. However, Boussouar et al. also found that lung CT abnormalities were not influenced by the types of COL3A1 variants. 71 Currently, there is no cure for vEDS. However, standard treatment for pneumothorax has been shown to be effective. 75
Loeys-Dietz syndrome
LDS is an autosomal dominant connective tissue disorder caused by mutations in genes encoding in the transforming growth factor beta- signaling pathway, such as TGFBR1, TGFBR2, or SMAD3. LDS is characterized by vascular findings (cerebral, thoracic, and abdominal arterial aneurysms/dissections) and skeletal manifestations (pectus excavatum or pectus carinatum, scoliosis, joint laxity, arachnodactyly, and talipes equinovarus). SP is an occasional feature of LDS. 76 A case report described SP as the presenting feature of a 26-year-old woman with this disease. 77
Others
Genetic syndromes associated with SP have other rarer etiologies than those described above, such as homocystinuria, 78 cutis laxa, 79 Sotos syndrome, 80 among others. 8 Homocystinuria is an autosomal recessive metabolic disorder caused by biallelic mutations in the gene encoding cystathionine β-synthase (CBS). Features are skeletal (tall stature with long limbs, scoliosis, pectus excavatum), ocular (lens dislocation, myopia), vascular (blood clots), central nervous system (intellectual disability, seizures), and cutaneous (hypopigmentation, livedo reticularis, malar flush). 78 Cutis laxa describes loose, redundant, hypoelastic skin, particularly over the neck, hands, groin, face, and trunk. Some types are inherited in an autosomal dominant or recessive manner. 79 Sotos syndrome is characterized by prenatal and postnatal increased growth, a variable learning disability, and characteristic facial features, mostly caused by functional loss of the NSD1 gene. 80 These syndromes are mostly reported as case reports of syndromes and lack epidemiological and clinical details pertaining to pneumothorax.
A diagnostic approach to patients with SP
Boone et al. propose that determining whether SP is caused by genetic disorders holds significant clinical implications for both patients and their relatives. 8 For the proband, establishing a genetic etiology can guide clinical decision-making regarding pneumothorax management, including assessing the necessity of pleurodesis after the first episode. Moreover, since genetically associated pneumothorax often presents with extrapulmonary complications, genetic diagnosis facilitates targeted surveillance and preventive measures, such as blood pressure monitoring and echocardiography in MFS, and renal tumor screening in BHD. For family members, regardless of whether they manifested pneumothorax symptoms, identification of pathogenic variants in the proband enables genetic testing, counseling, and surveillance/prevention strategies for relatives carrying the same variants. 8 Therefore, we recommend consideration of genetic etiologies in the routine evaluation of SP. Figure 4 illustrates the specific diagnostic pathway of SP, emphasizing the importance of a focused family history, consanguinity (which could heighten suspicion for an autosomal recessive condition), targeted history taking, and physical examination (including a thorough physical examination of the skin). When SP is suspected to be associated with a genetic syndrome, evaluation with chest HRCT is recommended. Based on the characteristics of pulmonary lesions, along with the aforementioned medical history and physical examination findings, the most likely matching genetic syndrome should be identified. Subsequently, collaboration with a clinical geneticist is advised to determine the necessity for further genetic testing and related investigations. Following a confirmed diagnosis, precise treatment and appropriate management can be implemented.

A diagnostic approach to patients with spontaneous pneumothorax.
The management of SP associated with genetic disorders diverges significantly from that of primary SP. Key distinctions include the recommendation of prompt genetic counseling, a more proactive approach to pleurodesis to mitigate high recurrence risk, and the implementation of a multidisciplinary team (MDT)-supported systemic management plan addressing extrapulmonary manifestations.
Conclusion
SP can result from various etiologies, often secondary to common respiratory diseases, but it may also indicate an underlying genetic syndrome. Clinicians should maintain a high index of suspicion for genetic syndromes, particularly in those with a positive family history. Patients with genetic syndrome-associated pneumothorax often present with extrapulmonary manifestations. Identifying a genetic cause has significant clinical implications, warranting a comprehensive evaluation that includes genetic counseling and a systematic MDT management approach. Due to the high recurrence rate of SP associated with genetic syndromes, active consideration of surgical intervention is recommended when indicated.