
Abstract
Background:
Respiratory diseases such as cystic fibrosis (CF), chronic obstructive pulmonary disease, and non-CF bronchiectasis are significant global health burdens. Current treatments aim to improve mucus clearance but do not fully address these diseases, highlighting the need for novel treatments. This study presents the results from phase I and phase IIb trials of GDC-6988, an inhaled, selective, and potent TMEM16A potentiator, in healthy volunteers.
Objectives:
To assess the safety and tolerability of orally inhaled GDC-6988 (nebulized and as a dry powder inhaler) in healthy subjects compared with placebo.
Design:
The phase I trial was a first-in-human, randomized, double-blind, placebo-controlled study to evaluate the safety, tolerability, and pharmacokinetics (PK) of single and repeat doses of nebulized GDC-6988.
Methods:
The study consisted of three parts: Part A with six cohorts (doses from 1.5 mg to 150 mg) using a single ascending dose (SAD) design; Part B with three cohorts (22.5 mg BID for 7 days, 75 mg BID for 14 days, and 45 mg BID for 14 days) using a multiple ascending dose (MAD) design; and Part C assessing the effect of salbutamol pretreatment on the highest dose tested in Part B (75 mg). The phase Ib study was a double-blind, randomized, placebo-controlled, single-center, multiple-dose escalation study evaluating the safety and PK of GDC-6988 DPI formulation, with and without salbutamol pretreatment. Three cohorts with doses of 11.2 mg, 28 mg, and 42 mg BID were tested. A bridging cohort compared PK in two capsule strengths of GDC-6988.
Results:
In the phase I study, 76 healthy subjects received GDC-6988 or placebo; in the phase Ib study, 41 subjects were enrolled (31 in MAD cohorts, 10 in the bridging cohort). GDC-6988 was safe and generally well tolerated, with no serious, severe, or grade ⩾3 adverse events observed at any dose level. Mild-to-moderate dose-dependent FEV1 declines were observed in both studies, but were mitigated by salbutamol pretreatment. In both trials, plasma PK concentrations of GDC-6988 were low, as expected.
Conclusion:
Inhaled GDC-6988 was safe and well tolerated across all dose levels. The plasma PK of GDC-6988 was low and generally dose-proportional with a relatively short half-life.
Trial registration:
Phase I: ClinicalTrials.gov Identifier NCT04488705; Phase Ib: ISRCTN30841680.
Introduction
Mucociliary clearance (MCC) is a key component of innate host defense that removes particles and pathogens from the airway and allows it to remain clear and unobstructed. 1 Effective mucus clearance from the airway is dependent upon mucus concentration and viscosity, which is largely affected by mucus hydration and mucin composition.2,3 Multiple ion channels expressed on secretory cells regulate fluid secretion into the airway to maintain mucus hydration. 3 Loss of effective MCC from disruption of mucus homeostasis or ciliary dysfunction can cause or contribute to muco-obstructive lung disease pathogenesis, as seen in cystic fibrosis (CF), non-CF bronchiectasis, and chronic obstructive pulmonary disease (COPD). 4 CF is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. 5 The present model of airway disease in CF is based on the concept that inadequate CFTR function results in reduced hydration of the airway mucosal surface. 6
TMEM16A (anoctamin-1, ANO1) is a calcium-activated chloride channel expressed at the apical surface of the airway epithelium that regulates chloride ion flow and subsequent airway surface hydration.7–11 Under inflammatory conditions, TMEM16A expression is enhanced. 12 Disease-associated inflammatory mediators such as IL-4, IL-13, and IL-6, as well as microbial virulence factors such as bacterial pyocyanin and lipopolysaccharide (LPS), have been associated with increased expression of TMEM16A in airway epithelial cells. 13 As TMEM16A is not mutated in CF, its potentiation provides a novel opportunity to “bypass” the genetic defect in CF, providing an alternate anion efflux pathway. Thus, TMEM16A potentiation may be able to improve mucus hydration and MCC, potentially addressing the clinical features of cough and excess sputum production often seen in patients with muco-obstructive diseases, thus reducing the downstream consequences of retained mucus such as chronic infection and acute exacerbations.
GDC-6988 (RO7506811; ETX001; ETD002) is an inhaled, selective, and potent TMEM16A potentiator. In vitro studies demonstrated that GDC-6988 increased airway surface hydration as well as increased the sensitivity of the TMEM16A ion channel to calcium, enhancing chloride ion flow within CF bronchial epithelial cells. 14 Additionally, in an in vivo study using a sheep model, GDC-6988 dose-dependently increased the MCC rate after a single dose with an effective dose in 100% population (ED100) of approximately 95 µg/kg. 14
A phase I, first-in-human study assessed the safety, tolerability, and pharmacokinetics (PK) of single ascending doses (SADs) and multiple ascending doses (MADs) of inhaled GDC-6988 delivered by nebulization in healthy volunteers (NCT04488705). Additionally, a phase Ib study assessed the safety, tolerability, and PK of SADs and MADs of inhaled GDC-6988 delivered by dry powder inhaler (DPI) in healthy volunteers, as well as comparing 2.8 mg and 7 mg capsules (ISRCTN30841680). Here, we report the results from both phase I studies.
Methods
These healthy volunteer studies were completed under two protocols: phase I and phase Ib. This manuscript follows the CONSORT (CONsolidated Standards Of Reporting Trials) statement. 15
Phase I
Study design
The phase I study (NCT04488705) was a first-in-human, randomized, double-blind, placebo-controlled study to assess the safety, tolerability, and PK of single and repeat doses of nebulized GDC-6988 in healthy volunteers. There were three parts in the study (Figure 1). Part A included six cohorts (doses from 1.5 mg to 150 mg) and utilized an SAD design; each cohort of eight subjects received a single dose of GDC-6988 or placebo in a 3:1 ratio (active:placebo). Part B included three cohorts (22.5 mg twice daily (BID) 7 days, 75 mg BID 14 days, and 45 mg BID 14 days) of eight subjects in an MAD design: each cohort received a BID dose of GDC-6988 or placebo in a 3:1 ratio (active:placebo) for 7 days (Cohort 9) or 14 days (Cohorts 10 & 11). Part C examined the effect of salbutamol pretreatment on the highest dose tested in Part B (i.e., 75 mg): each cohort received a 75 mg BID dose of GDC-6988 or placebo in a 3:1 ratio (active:placebo) for 7 days, with subjects receiving pretreatment with BID doses of salbutamol on Days 5–7; each cohort consisted of eight subjects. All cohorts were dosed sequentially.

Phase I study design.
Key inclusion and exclusion criteria
Male and female subjects aged 18–55 years, with a body weight of ⩾50 kg and body mass index (BMI) within the range of 19–30 kg/m2, were included in the study. Individuals with upper or lower respiratory tract infection within 4 weeks of the screening visit or randomization were excluded. A full list of inclusion and exclusion criteria can be found in the Supplemental Methods.
Study objectives and endpoints
The primary objectives were to assess the safety and tolerability of increasing single doses and repeat doses of nebulized GDC-6988 in healthy subjects compared with placebo, and, in Part C, to assess the safety of repeat doses of inhaled GDC-6988 or placebo in healthy subjects when pretreated with inhaled salbutamol. Secondary objectives included determining the PK parameters of GDC-6988 following increasing single and repeat doses of inhaled GDC-6988, and in Part C, to monitor and determine PK parameters following repeated dosing of inhaled GDC-6988 with and without pretreatment of inhaled salbutamol.
Safety assessments
Safety and tolerability of GDC-6988 were evaluated throughout the study by review of physical examination, vital signs (blood pressure, pulse rate, respiration rate, temperature, and oxygen saturation), electrocardiogram (ECG) assessments, clinical laboratory testing, spirometry measurements (forced expiratory volume in 1 second (FEV1)), assessment of AEs, and use of concomitant medications.
PK assessments
Plasma PK samples were collected on Days 1–4 (Part A), Days 1–8 or Days 1–15 (Part B), and Days 1–8 (Part C), depending on the cohort assigned. GDC-6988 plasma concentrations were analyzed using a validated, specific, and sensitive method with a lower limit of quantification (LLOQ) of 0.05 ng/mL. PK parameters in plasma for GDC-6988 were calculated using non-compartmental analysis (NCA). Key parameters include maximum observed plasma concentration (Cmax), time to Cmax (Tmax), AUC for a dose interval (AUC0-12), AUC from time zero to infinity (AUC0-inf), apparent clearance (CL/F), apparent central volume of distribution (Vz/F), and apparent terminal half-life (t1/2).
Spirometry
Spirometry assessments were performed up to eight times to obtain two reproducible readings according to the American Thoracic Society (ATS)/European Respiratory Society (ERS) guidelines. 16 The highest FEV1 and forced vital capacity (FVC) readings from each assessment were used for analysis (including for calculation of the FEV1/FVC ratio) even if the FEV1 and FVC values came from two different forced exhalations. Spirometry assessments were performed in accordance with ATS/ERS guidelines, and predicted values were calculated using the reference equations in the National Health and Nutrition Examination Survey reference equations. 17
Statistical analysis
Descriptive statistics (e.g., mean, geometric mean, median, standard deviation, coefficient of variation) were deemed sufficient for the study’s objectives. The sample size was determined based on feasibility rather than statistical considerations. The safety population was defined as all subjects who received at least one dose of study medication. The PK population was defined as subjects in the safety population who provided at least one evaluable sample result for the determination of PK.
Phase Ib
Study design
The phase Ib study was a double-blind, randomized, placebo-controlled, single-center, multiple-dose escalation study that evaluated the safety and pharmacokinetics of MAD of GDC-6988 DPI formulation without and with pretreatment with salbutamol in healthy adult subjects (Figure 2). Three cohorts with doses of 11.2 mg, 28 mg, and 42 mg BID were tested in the study. Within each MAD cohort, 10 subjects were randomly assigned at a 4:1 ratio to receive GDC-6988 or placebo twice daily for 14 days. Subjects received pretreatment with salbutamol (200 µg) 15 (±5) minutes prior to administration of the study drug, starting from Day 8 for subjects in Cohort 1 (11.2 mg BID) and Cohort 2 (28 mg BID) and from Day 1 for subjects in Cohort 3 (42 mg BID). Sentinel dosing was employed with the first two subjects in all MAD cohorts.
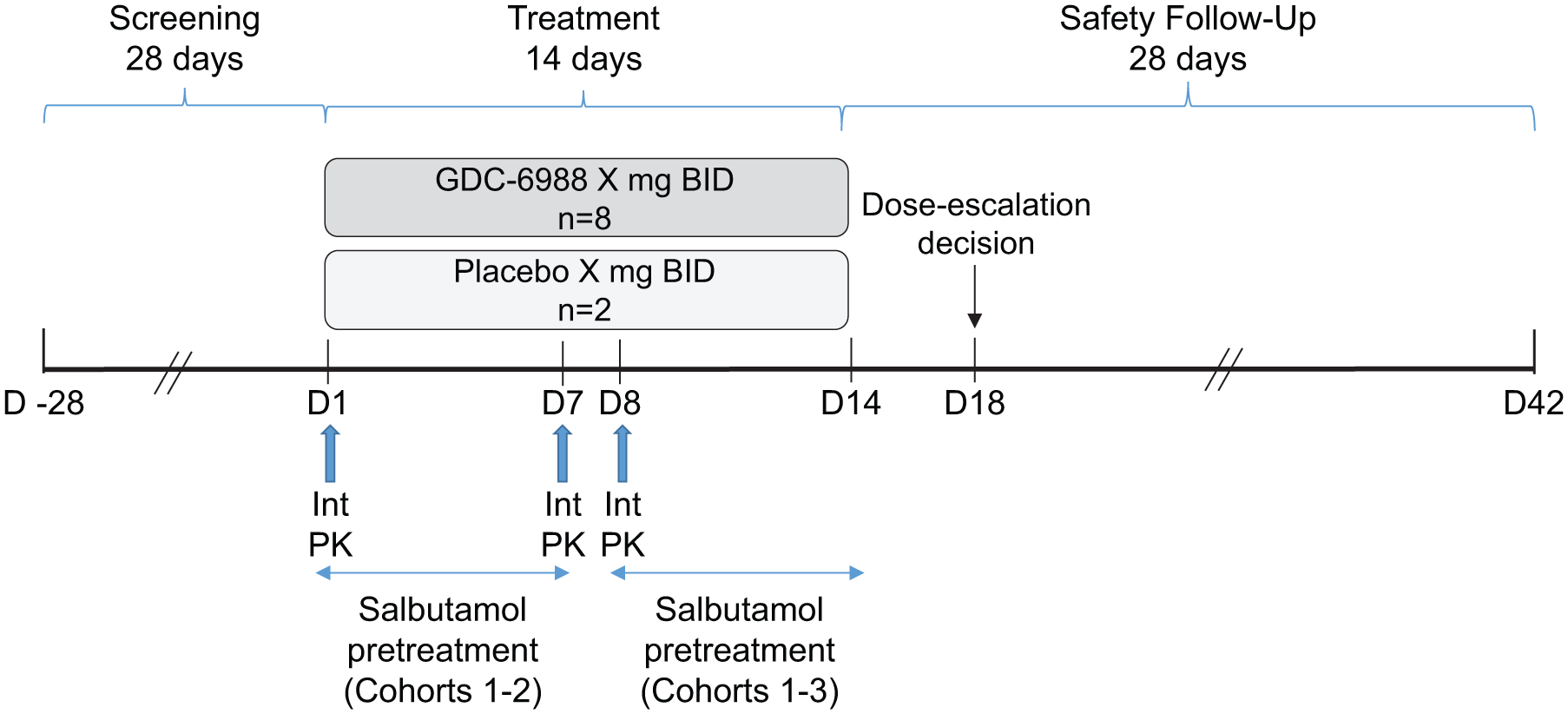
Phase Ib study design.
This study also included a bridging cohort to compare the plasma PK and safety profiles of equivalent nominal doses of 42 mg achieved with 2.8-mg capsules compared with 7-mg capsules (Supplemental Figure 1). Within this bridging cohort, 10 subjects were randomly assigned at a 4:1 ratio to receive 42 mg GDC-6988 or placebo. On Day 1, subjects received a single dose of GDC-6988 or placebo using 2.8-mg capsules (i.e., 15 capsules); on Day 8, subjects received an equivalent single dose (e.g., 42 mg) of GDC-6988 or placebo using 7-mg capsules (i.e., 6 capsules).
All active and placebo doses in all cohorts were administered via oral inhalation using the Smart DPI, a customized version of the commercially available Plastiape RS01 Monodose DPI (Plastiape S.p.A, Italy).
Key inclusion and exclusion criteria
Male or female subjects aged 18–55 years with a BMI within the range of 18–32 kg/m2 were included in the study. A full list of inclusion and exclusion criteria for the phase Ib study can be found in the Supplemental Methods.
Study objectives and endpoints
The primary objective was to evaluate the safety of GDC-6988 as delivered via the Smart DPI. Secondary objectives were to characterize the PK of GDC-6988 as delivered via the Smart DPI, evaluate the performance of the Smart DPI device, and identify a recommended phase II dose and regimen for GDC-6988. The exploratory objectives were to evaluate potential relationships between drug exposure and the safety of GDC-6988, to assess the palatability of GDC-6988 administered via DPI, and to compare the safety and PK of 2.8-mg and 7-mg GDC-6988 capsules as delivered via the Smart DPI device.
Safety assessments
Safety assessments consisted of monitoring and recording AEs, including SAEs, and AEs of special interest, laboratory assessments, vital signs, electrocardiogram (ECG) assessments, clinical laboratory testing, spirometry measurements (forced expiratory volume in 1 second (FEV1)), and other protocol-specified tests deemed critical to the safety evaluation of the study.
PK assessments
Plasma PK samples were obtained on Days 1, 2, 7-9, 14, 15, 18, and 42 for the MAD cohorts; for the bridging cohort, plasma PK samples were obtained on Days 1, 2, 8, 9, and 22. PK parameters included but were not limited to: maximum observed plasma concentration (Cmax), time to Cmax (tmax), AUC from time zero to infinity (AUC0-inf), AUC from time zero to the sampling time at 12 h (AUC0-12), apparent clearance (CL/F), apparent central volume of distribution (Vz/F), and apparent terminal half-life (t1/2). The LLOQ for the plasma assay is 0.02 ng/mL.
Spirometry and oscillometry assessments
Spirometry was conducted during the study in conjunction with other assessments. Forced oscillometry technique was based on the ERS Task Force on Respiratory Impedance Measurements, 18 with measures including resistance at low frequency, 5 Hz (R5), resistance change (R520), AX, and reactance difference inspiration – expiration (X5). Oscillometry was conducted immediately before spirometry when both were performed during the same time window.
Patient-reported outcomes
A single-item patient-reported outcome instrument was completed to assess the palatability of GDC-6988 DPI.
Statistical analysis
Statistical summaries were descriptive in nature (e.g., mean, geometric mean, standard deviation, median, and range for continuous variables and counts and percentages for categorical variables). Subjects were grouped according to treatment received, and any subjects who received any amount of GDC-6988 or placebo were included in the analysis. All subjects assigned to placebo (regardless of dose group) were combined into a single placebo control group. The safety analysis population consisted of all subjects who received at least one dose of the study drug. The PK analysis population consisted of subjects receiving active drug with sufficient data to enable estimation of key parameters (e.g., AUC, Cmax, half-life).
Results
Phase I
In total, 76 healthy subjects received GDC-6988 or placebo (Table 1). In Part A, the mean age of subjects was 33.5 years; most subjects were male (83.3%) and most subjects were White (75.0%). In Part B, the mean age of subjects was 32.9 years; most subjects were male (87.5%) and most subjects were White (79.2%). In Part C, the mean age of subjects was 35.8 years; most subjects were male (87.5%) and most subjects were White (62.5%).
Phase I subject demographics and baseline disease characteristics.
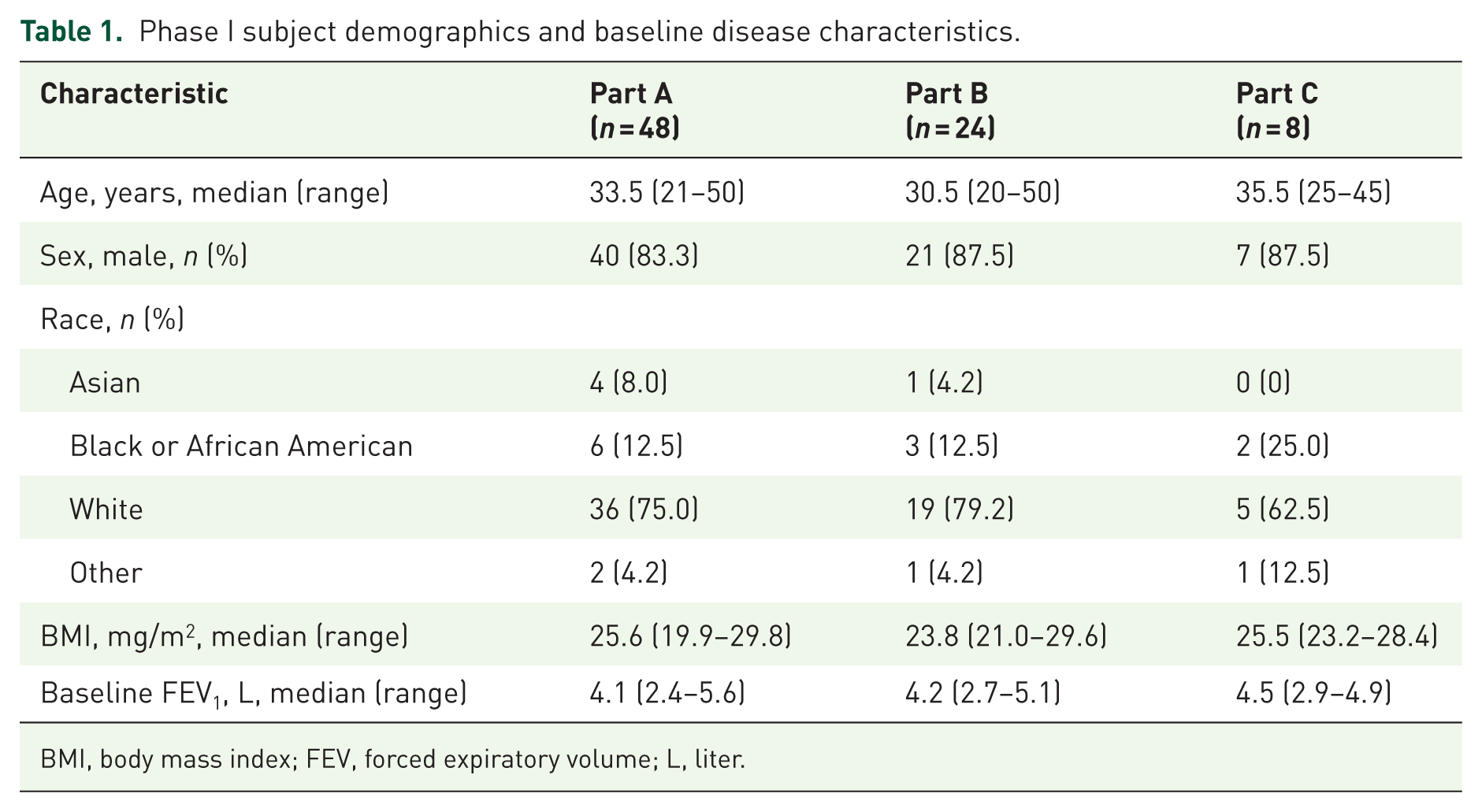
BMI, body mass index; FEV, forced expiratory volume; L, liter.
Safety
In Part A, 10 (20.8%) subjects reported 11 treatment-emergent AEs (TEAEs), of which one was considered by the investigator to be study drug-related. None of the TEAEs were serious and there were no TEAEs leading to subject withdrawal. All TEAEs were mild to moderate in severity. The most frequently reported TEAE was headache, with no other TEAE reported by more than one subject (Supplemental Table 1). There were no clinically significant changes in vital signs, clinical chemistry or hematology parameters, or physical examinations.
In Part B, 12 (50.0%) subjects experienced 26 TEAEs, of which 5 were considered by the Investigator to be drug-related. The most common TEAEs were headache (n = 4) and contact dermatitis (n = 2; Supplemental Table 2). All TEAEs in Part B were mild or moderate. Two subjects in Part B experienced changes in spirometry parameters which led to study withdrawal due to protocol-defined stopping criteria. One subject in the 75 mg cohort experienced moderate, intermittent chest tightness “probably related” to study medication and a decrease in the FEV1 value >15% from baseline on Study Day 6 resulting in treatment withdrawal. Another subject in the 75 mg cohort experienced a consistent drop in FEV1 of >20% post-dose on Study Day 12 and was withdrawn from treatment.
In Part C, 2 (25.0%) subjects experienced a total of 2 TEAEs, one of which was considered by the Investigator to be study drug-related (wheezing, reported by a subject in the 75.0 mg of GDC-6988 BID for 7 days cohort); both TEAEs were mild in severity, and neither was serious nor led to subject withdrawal (Supplemental Table 3). There were no clinically significant findings, vital signs, ECG, or laboratory changes reported.
Spirometry
In Part A, subjects had a decrease from baseline in FEV1 30 minutes following the administration of GDC-6988 that was resolved 2 h after administration; these changes were not considered clinically significant and were not associated with any TEAEs (Supplemental Figure 2). A similar, transient mean decrease from baseline FEV1 of 0.2 to 0.3 L was also observed in Part B of the study, which was most clearly observed in the 45 mg BID and 75 mg BID groups (Supplemental Figure 3). In Part C, FEV1 variations were again observed on Days 1–4; however, salbutamol 200 µg pretreatment on study Days 5–7 mitigated the FEV1 decreases seen in Parts A and B of the study (Figure 3). There was no apparent correlation between systemic exposure of GDC-6988 and change in FEV1.

Pre- to post-dose FEV1 difference with and without salbutamol pretreatment in Part C.
Pharmacokinetics
The mean concentration–time profiles of the single doses tested in the SAD cohorts are shown in Figure 4(a). The PK parameters from the SAD cohorts are displayed in Table 2. Following single oral inhalation dosing, GDC-6988 was rapidly absorbed, with the median time to maximum observed plasma concentration (Tmax) occurring between 0.083 h (5 min) and 0.167 h (10 min) post-dose across the dose range. Over the dosing range of 1.5 and 150 mg GDC-6988, the geometric mean Cmax increased from 2.09 ng/mL to 93.4 ng/mL, and the geometric mean AUC0-inf increased from 0.904 to 86.2 ng*h/mL. These increases were approximately dose-proportional, with the Cmax slightly less than dose-proportional, which is expected since inhalation times are variable across doses (i.e., higher dose levels having longer nebulization duration). The intersubject variability observed upon single dosing associated with exposure-related parameters was considered moderate to high, ranging from 13.5% to 98.7%.

Mean pharmacokinetic profiles of nebulized GDC-6988 in healthy volunteers in (a) Part A SAD, (b) Part B MAD, and (c) Part C. BID, twice daily.
Plasma PK parameters for GDC-6988 in Parts A–C of phase I.
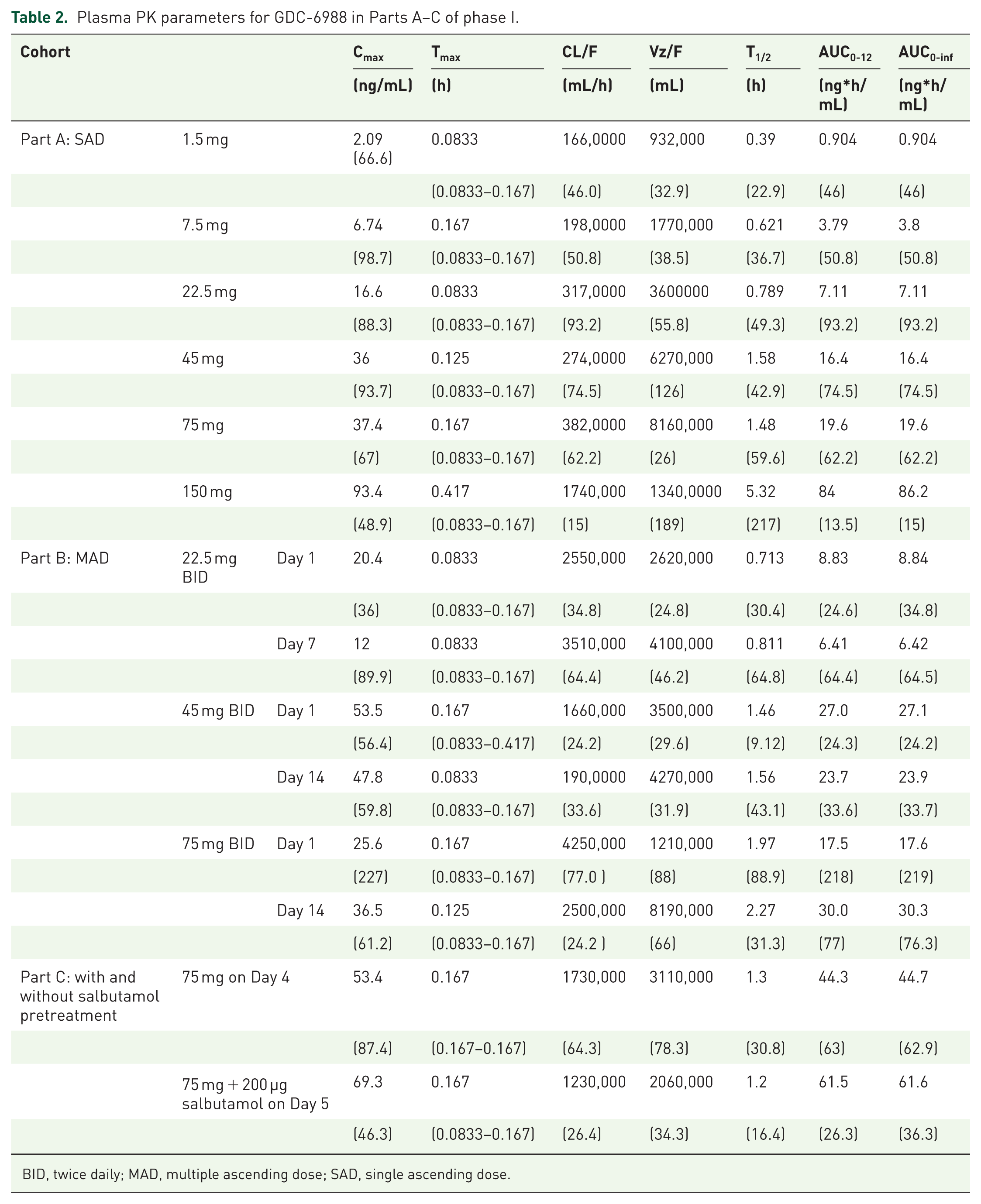
BID, twice daily; MAD, multiple ascending dose; SAD, single ascending dose.
The mean concentration–time profiles of the three MAD cohorts studied in Part B are shown in Figure 4(b), and Table 2 lists the multiple-dose PK parameters. GDC-6988 was rapidly absorbed following oral inhalation. At the highest administered dose of 75 mg BID on Day 14, the geometric mean Cmax was 36.5 ng/mL, and the geometric mean AUC0-12 (AUC) was 30.0 ng*h/mL, and the variability associated with exposure-related parameters at steady state was considered relatively high at between 61.2% and 77.0% CV. Due to the rapid systemic clearance, no considerable accumulation of GDC-6988 was observed upon repeat dosing.
In study Part C, the Cmax and AUC geometric mean ratios (geometric CV%) between administration with salbutamol (Day 5) and without salbutamol (Day 4) were 1.3 (34.5%) and 1.38 (38.6%), respectively. Overall, a comparison of the exposure-related parameters without and with salbutamol showed no clear difference, other than perhaps a reduced variability observed in this group on Day 5, where salbutamol was coadministered (Figure 4(c)). PK parameters in Part C are listed in Table 2.
Phase Ib
A total of 41 subjects were enrolled (31 subjects were assigned to MAD cohorts, and 10 subjects were assigned to the bridging cohort) (Table 3). In the MAD cohorts, the median age was 40 years (range: 24–53); most subjects were male (71.0%) and most subjects were White (83.9%). The median percentage predicted FEV1 (ppFEV1) at baseline was 97.2 (range: 81.9–127.3). In the bridging cohort, the median age was 45.5 years (range: 27–53); half of the subjects were male, and most subjects were White (80%). The median ppFEV1 at baseline was 110.3 (range: 86.9–137.5).
Phase Ib subject demographics and baseline disease characteristics.

BMI, body mass index; ppFEV, percent predicted forced expiratory volume.
A total of three subjects (7.3%) discontinued study treatment; two subjects withdrew themselves, and one subject discontinued treatment due to an AE. Across all the MAD cohorts, the median treatment duration was 14 days, and the median number of administered doses was 28. In the bridging cohort, the median number of administered doses was 2, each separated by 7 days. The median cumulative dose was 84.0 mg among subjects receiving GDC-6988.
Safety
There were 3 (37.5%), 6 (75.0%), and 6 (75.0%) subjects in the 11.2 mg, 28 mg, and 42 mg GDC-6988 cohorts, respectively, who experienced ⩾1 AE; 4 (57.1%) placebo subjects experienced ⩾1 AE. The most common AEs (reported in at least two subjects) were headache, back pain, and throat tightness (Supplemental Table 4). One subject in the 42 mg cohort experienced a Grade 1 AE of special interest of supraventricular tachycardia, which was assessed as related to both blinded GDC-6988 and salbutamol (as described below), leading to treatment discontinuation. No AEs led to dose modification or interruption in any of the MAD cohorts. No Grade ⩾3 AEs, SAEs, or deaths were reported in any of the subjects in the MAD cohorts, and no subjects were withdrawn from the study due to an AE. Among subjects receiving GDC-6988, 1 subject (12.5%) in the 11.2 mg cohort, 4 subjects (50%) in the 28 mg cohort, and one subject (12.5%) in the 42 mg cohort experienced an AE assessed as related to GDC-6988 or placebo; additionally, one subject on placebo experienced an AE assessed as related to GDC-6988 or placebo. The AE assessed as related to GDC-6988 in the 42 mg cohort subject was also assessed as related to salbutamol. In the bridging cohort, 1 subject receiving GDC-6988 (12.5%) experienced an AE of Grade 1 diarrhea; none of the subjects in the placebo group experienced an AE. There were no Grade 3–4 AEs, SAEs, or deaths reported in the bridging cohort. Additionally, none of the subjects were withdrawn from the study due to an AE, and none of the AEs led to dose modifications or interruptions. None of the subjects in the bridging cohort experienced any related AEs.
Spirometry
Without salbutamol pretreatment, a mean decrease in FEV1 of approximately 2.5% and 5.0% was observed 30 minutes after administering GDC-6988 to subjects in the 11.2 mg and 28 mg BID cohorts, respectively, compared with no obvious change observed in the placebo subjects (Figure 5). Mean FEV1 trended back to the daily baseline 2 h after treatment. With salbutamol pretreatment, the negative changes from the daily baseline in mean FEV1 were mitigated, with a positive trend observed for subjects on the 11.2 mg and 28 mg BID GDC-6988 and the placebo subjects. In subjects receiving GDC-6988 42 mg BID, salbutamol pretreatment was used on all treatment days. In the bridging cohort, where all subjects were pretreated with salbutamol prior to GDC-6988, there was no significant change in FEV1 from baseline.

Mean percent change in FEV1 from daily baseline.
Oscillometry
On Day 3, an increase from the daily baseline in oscillometry parameters of resistance at 5 Hz, resistance at 5 Hz to 20 Hz, resonance frequency, and reactance area, and a decrease from the daily baseline in reactance at 5 Hz were observed at 30 (5) min after the morning doses in subjects receiving GDC-6988 11.2 mg BID and 28 mg BID without salbutamol pretreatment. Oscillometry parameters trended back to the daily baseline 6 h after treatment. In subjects on GDC-6988 42 mg BID, salbutamol pretreatment was used on all days, and no clinically significant changes in oscillometry parameters were observed.
Palatability of GDC-6988
The majority of subjects reported a “slightly pleasant” (6 subjects; 21.4%) or “barely detectable” (10 subjects; 35.7%) taste; 2 subjects (7.1%) reported a “slightly unpleasant” taste.
Pharmacokinetics
Following the initial oral inhaled dose of GDC-6988 11.2, 28, or 42 mg BID on Day 1, Day 7, and Day 8 in the MAD cohorts, the geometric mean maximum concentration (Cmax) exhibited an approximately dose-proportional increase with Tmax between 0.08 to 0.7 h post-dose, followed by a rapid decline of the concentrations (Figure 6(a)). PK parameters are summarized in Table 4.

Mean (±SD) plasma concentration–time profiles of (a) GDC-6988 in MAD cohorts 1–3 and (b) in the bridging cohort.
Plasma PK parameters for GDC-6988 in MAD cohorts and bridging cohort.

BID, twice daily; MAD, multiple ascending dose.
The AUC0-12 similarly demonstrated an approximately dose-proportional increase. The coefficient of variation (CV%) for both Cmax and AUC ranged from 15.9% to 51.9%. The geometric mean half-lives were consistent across all cohorts with a range of 1.7 to 2.4 h. The 11.2 and 28 mg cohorts did not show distinct PK accumulation at steady state; this finding can be attributed to the short half-life of the drug (around 2 h). Additionally, at steady state, the exposures on Day 8, when the drug was administered with salbutamol, were slightly higher (approximately 10%–35%) compared to Day 7, when the drug was administered without salbutamol. However, no clear difference in PK variability was observed between Day 8 (with salbutamol) and Day 7 (without salbutamol). In the 42 mg BID cohort, a slight PK accumulation (approximately 29% in AUC0-12) at steady state was observed; this accumulation is likely attributed to the PK variability observed within this cohort.
In the bridging cohort (Figure 6(b)), slightly higher Cmax and AUC values were observed on Day 8, when a single dose of 42 mg with 7-mg capsules was administered, compared to Day 1, when a single dose of 42 mg with 2.8-mg capsules was used. Specifically, there was an approximate 8% increase in Cmax and an 18% increase in AUC; however, given the PK variability associated with the 42 mg dosing regimen, the marginal increase in exposure might solely be attributed to PK variability.
Discussion
GDC-6988 is a selective and potent TMEM16A potentiator, which may improve mucus hydration and mucociliary clearance in patients with muco-obstructive diseases. Both the phase I and phase Ib studies of GDC-6988 demonstrated that GDC-6988 was safe and generally well tolerated when administered via nebulization and via a DPI, with no serious, severe, or grade ⩾3 AEs observed across all dose levels evaluated. Safety findings were relatively consistent across studies, including dose-dependent FEV1 declines observed in both studies. The FEV1 declines generally peaked around 30 min after administration and typically returned to baseline by 2 h after administration, which is consistent with the relatively short half-life of GDC-6988. Notably, these declines in FEV1 were largely asymptomatic or minimally symptomatic; however, two patients in Part B of the phase I study who were not pretreated with salbutamol were withdrawn due to changes in spirometry parameters. Administering salbutamol as a pretreatment mitigated the declines in FEV1 in both studies, and the bronchodilatory effects of salbutamol exceeded the bronchoconstriction effects of GDC-6988, such that following salbutamol premedication and GDC-6988 administration, FEV1 was above baseline. It is impossible to know for certain whether this represents an on-target effect of GDC-6988, though the timing of the FEV1 changes, which aligns with the pharmacokinetic characteristics of GDC-6988, suggests that this does not represent a nonspecific response to airway irritation. Although GDC-6988 (then known as ETX-001) was tested in freshly isolated human bronchi in vitro and did not have any effect on the contractile or relaxant responses of the smooth muscle, 19 direct agonism of TMEM16A in human airway smooth muscle induced significant contraction, raising the possibility that the observed declines in FEV1 represent TMEM16A potentiation via GDC-6988 on airway smooth muscle. 20 It is unknown why the experimental conditions of the in vitro model of GDC-6988 did not predict the observed human effect on FEV1.
Overall, no serious or severe AEs were observed throughout either study. However, one subject experienced an AE of special interest of supraventricular tachycardia, observed on Holter monitor, which led to treatment discontinuation. The subject was asymptomatic during the event, and the dysrhythmia resolved without intervention. The event was considered related to both salbutamol and blinded GDC-6988, and since the event occurred shortly after the administration of both salbutamol and study drug, it is impossible to know which, if any, of the two drugs was responsible for the event. Treatment with salbutamol is known to be associated with supraventricular tachycardias, 21 and although systemic exposures of GDC-6988 were low, TMEM16A is expressed in atrial fibroblasts. 22
In both phase I and phase IIb studies, the observed plasma PK concentrations of GDC-6988 were generally low, as expected. The effective half-life for systemic PK was also relatively short (around 2 h), resulting in no clear PK accumulation in the BID multiple dosing cohorts. The salbutamol pretreatment had a very small impact on the PK profiles with GDC-6988 mean PK profiles slightly higher with salbutamol pretreatment than those without pre-treatment, and a large overlap in the variability bands. This is unlikely to be due to PK drug–drug interaction as both molecules were orally inhaled with low systemic exposure, and automated whole-cell planar patch-clamp electrophysiology studies also confirmed minimal pharmacodynamic drug–drug interaction (i.e. salbutamol does not modulate human TMEM16A currents or affect the response of human TMEM16A currents to GDC-6988) (data on file). The small difference noted may be due to slight changes in the regional lung deposition, given the bronchodilation effect of salbutamol.
The phase Ib DPI study was conducted after the phase I nebulized study because the nebulized suspension formulation of GDC-6988 was considered unstable and therefore would not support dosing GDC-6988 in a home setting. The phase Ib DPI dose selection was based on PK and safety data from the phase I nebulized study and the nonclinical toxicology studies with the DP formulation (data on file). The starting dose of GDC-6988, 11.2 mg BID, is two times the approximate therapeutic lung dose estimated based on the preclinical model, 14 and predicted to be equivalent to the deposited lung dose for the 45 mg BID nebulized formulation (calculated based on the fine particle fraction from in vitro study), a dose found to be safe and well tolerated in the MAD part of the phase I nebulization study. Based on the observed plasma PK data, compared to the nebulized 45 mg BID dose, the 11.2 mg BID DPI dose measured on Day 7, without salbutamol, had a lower steady-state mean Cmax (25.8 ng/mL (CV% 24.9%) vs 47.8 ng/mL (CV% 59.8%)) and higher steady-state AUC0-tau (33.6 ng*h/mL (CV% 27.3%) vs 23.7 ng*h/mL (CV% 33.6%)). Given that the systemic PK properties (e.g. metabolism and distribution) of GDC-6988 are expected to be the same in both formulations, the differences in the PK parameters and profiles between nebulized 45 mg BID and DPI 11.2 mg BID (assumed to have equivalent lung dose) are likely due to the regional lung deposition differences in the two formulations/devices and also the translatability of the in vitro lung dose estimate, based on fine particle fraction, to the actual lung dose in human subjects. To explore the lung deposition difference between these two formulations/devices, a mechanistic lung physiologically based pharmacokinetic (PBPK) model, accounting for lung deposition, lung and oral absorption, systemic clearance, and tissue distribution, was built, refined, and verified with GDC-6988 plasma PK data from the phase I and phase Ib studies, 23 thus confirming that there is a clear difference in the regional lung deposition between nebulized suspension and DP formulation/device, which can contribute to the PK profile differences. The bridging cohort in the phase Ib study confirmed similar PK profiles between the 42 mg dose with 2.8-mg capsules and with 7-mg capsules, which supports the usage of 7-mg capsules in future clinical studies to decrease the capsule burden for patients.
Some potential limitations of these studies included the limited treatment duration of 14 days. Furthermore, these studies were conducted in healthy volunteers, and the results may not extend to patients with respiratory disorders. In particular, it is unknown how the changes in FEV1 will affect patients with obstructive lung disease. This limitation is currently being tested in the ongoing study NCT06603246, where a safety run-in cohort is being dosed with low and high doses of GDC-6988 without and with salbutamol, to ensure that GDC-6988 can be safely administered to this population, with appropriate premedication, if needed. Additionally, while the phase Ib study closely resembled the UK population, this may not extrapolate to more diverse global populations, and the in-clinic treatment may not translate to real-world conditions.
Conclusion
Inhaled GDC-6988 was considered safe and well tolerated across all dose levels. The plasma PK of inhaled GDC-6988 was low and generally dose-proportional with a relatively short half-life. A phase Ic biomarker study (NCT06603246) is currently underway.
Supplemental Material
sj-docx-1-tar-10.1177_17534666251393346 – Supplemental material for Randomized, phase I studies to evaluate the safety, tolerability, and pharmacokinetics of an inhaled, TMEM16A potentiator, GDC-6988, in healthy subjects
Supplemental material, sj-docx-1-tar-10.1177_17534666251393346 for Randomized, phase I studies to evaluate the safety, tolerability, and pharmacokinetics of an inhaled, TMEM16A potentiator, GDC-6988, in healthy subjects by Paul Miller, Daniel Repplinger, Rui Zhu, Yiling Chen, Nicholas Lewin-Koh, David Morris, Paul Russell, Gaohong She, Nand Singh, Rachael White, Denisa Wilkes, Hubert Chen and Joshua Galanter in Therapeutic Advances in Respiratory Disease
Supplemental Material
sj-docx-2-tar-10.1177_17534666251393346 – Supplemental material for Randomized, phase I studies to evaluate the safety, tolerability, and pharmacokinetics of an inhaled, TMEM16A potentiator, GDC-6988, in healthy subjects
Supplemental material, sj-docx-2-tar-10.1177_17534666251393346 for Randomized, phase I studies to evaluate the safety, tolerability, and pharmacokinetics of an inhaled, TMEM16A potentiator, GDC-6988, in healthy subjects by Paul Miller, Daniel Repplinger, Rui Zhu, Yiling Chen, Nicholas Lewin-Koh, David Morris, Paul Russell, Gaohong She, Nand Singh, Rachael White, Denisa Wilkes, Hubert Chen and Joshua Galanter in Therapeutic Advances in Respiratory Disease
Footnotes
Acknowledgements
The authors wish to thank all the participants, their families, and the investigators who participated in this study. Writing assistance provided by Lindsey Kirkland, PhD (Genentech, Inc.).
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.