
Abstract
We report a case of tracheobronchial amyloidosis (TBA) in a 55-year-old woman with newly diagnosed primary Sjögren’s syndrome (SS), presenting with persistent cough, hemoptysis, and dry mucosal symptoms. Chest CT showed thickened airway walls and cystic lung changes, while bronchoscopy revealed nodular lesions with exposed vessels. Congo red staining confirmed amyloid deposition with κ light-chain dominance, consistent with AL amyloidosis. Despite immunosuppressive therapy, airway lesions persisted, highlighting the challenge of managing localized amyloidosis in SS. This case underscores the need for early recognition of TBA in SS patients presenting with respiratory symptoms.
Introduction
Tracheobronchial amyloidosis (TBA) is a rare localized form of AL amyloidosis, characterized by progressive airway involvement leading to structural damage and respiratory symptoms.1,2 Sjögren’s syndrome (SS) is an autoimmune disorder primarily affecting exocrine glands, but may also present with pulmonary complications, including interstitial lung disease, lymphoma, and amyloidosis.3,4 Although amyloidosis is a recognized but rare complication of SS, isolated airway involvement remains exceedingly uncommon. This case highlights the diagnostic and therapeutic challenges of TBA in SS, emphasizing the need for early recognition to avoid misdiagnosis.
We have followed the relevant CARE guidelines (Supplemental Material). 5 The mandatory items of the CARE guidelines include: title, keywords, abstract, case presentation, patient information, clinical findings, diagnostic assessment, therapeutic intervention, outcome and follow-up, discussion, patient perspective, informed consent, and ethics statement.
Case presentation
Initial presentation
The patient was a 55-year-old woman with a medical history of hyperthyroidism who presented with acute symptoms, including cough, sputum production, and hemoptysis (blood-streaked sputum) that had persisted for 6 days. She was also experiencing dry eye and dry mouth. She had no history of smoking or recurrent infections and had not been previously diagnosed with Sjögren’s syndrome. Upon hospital admission, hemostatic agents, including etamsylate, tranexamic acid, and carbazochrome, were administered to control the bleeding. In addition, empirical anti-inflammatory treatment was initiated with ceftizoxime. Hemoptysis did not recur after the treatment. Furthermore, the patient did not show pulmonary hypertension or airway stenosis.
Imaging findings
Computed tomography (CT) revealed increased thickness of the airway wall in both the right intermediate bronchus and the left lower lobe bronchus (Figure 1). Airway amyloidosis exhibits nonspecific and variable patterns. CT also showed the presence of cystic shadows distributed over the entire lung (Figure 2), a characteristic of SS. Nodular lesions were identified in the right intermediate bronchus and the left lower lobe bronchus, and exposed vessels could be observed at these lesion sites (Figure 3). Subsequently, a mucosal biopsy of the right intermediate bronchial nodule was performed under bronchoscopy.
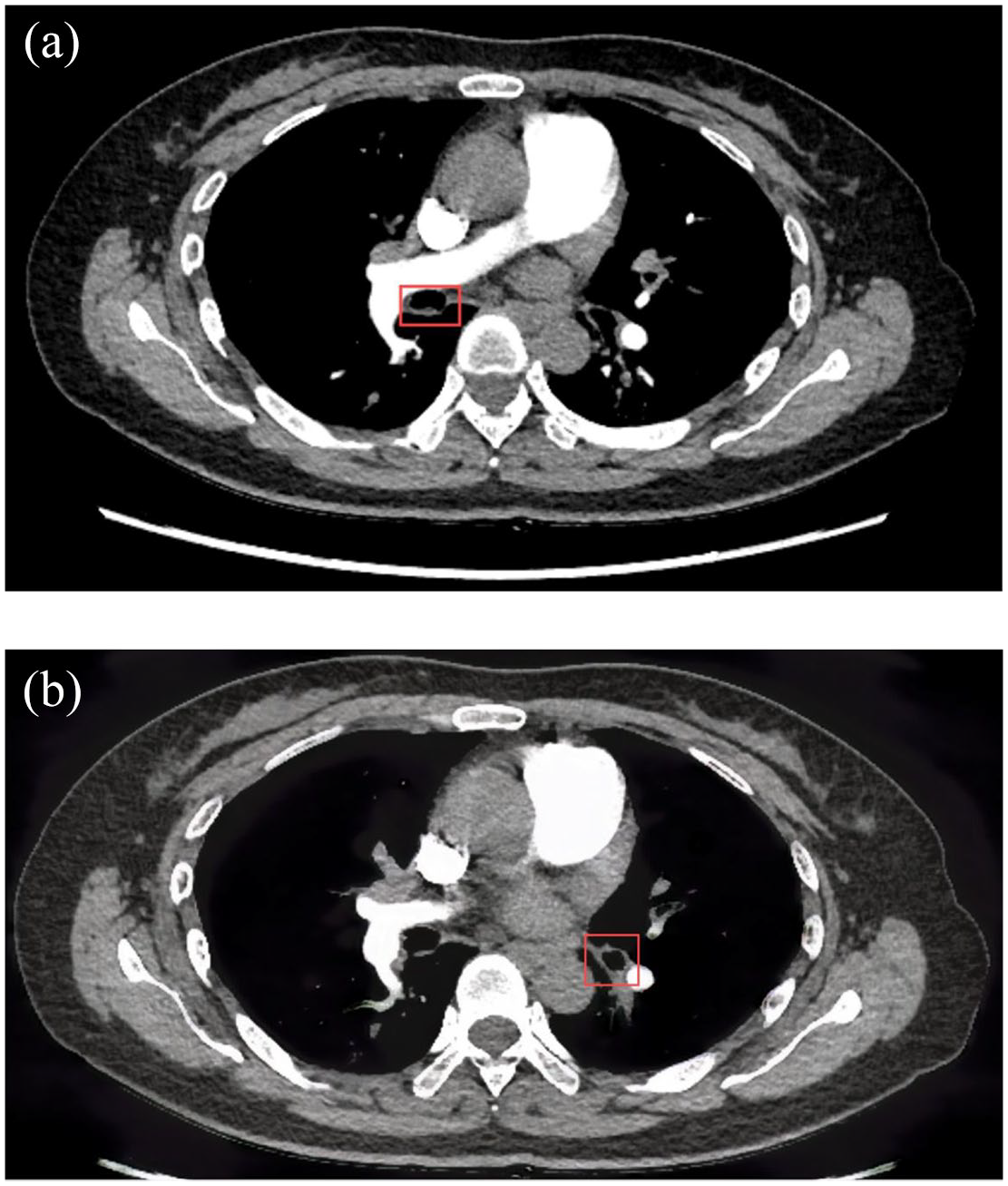
The CT manifestations in a patient with tracheobronchial amyloidosis and Sjögren’s syndrome (focus on lesions in bronchus). (a) lesions in the right intermediate bronchus, (b) lesions in left lower lobe bronchus.

The CT manifestations in a patient with tracheobronchial amyloidosis and Sjögren’s syndrome (focus on the characteristics of Sjögren’s syndrome). (a) cystic shadows over the right and left upper lobes, (b) cystic shadow over the left upper lobe; (c): cystic shadow over the right middle lobe; (d) cystic shadows over the right lower lobe.

Bronchoscopy findings in a patient with airway amyloidosis. (a) lesions in the right intermediate bronchus (b, c) lesions in the left lower lobe bronchus.
Serological testing
In addition, the levels of inflammatory markers were not elevated: C-reactive protein, 1.5 mg/L; serum amyloid A (SAA), <4.8 mg/L; procalcitonin (PCT), <0.02 ng/mL; and white blood cell count, 4.90 × 109/L. However, the patient had an elevated rheumatoid factor level (98.2 IU/mL) and a high titer of antinuclear antibodies (1:320); she also tested positive for both anti-SSA and anti-SSB antibodies. These immunological markers are consistent with SS. Furthermore, the patient had been experiencing dry mouth and eyes for over 10 years. Based on these findings and those of a pathological examination of the soft tissue of the lip mucosa, a diagnosis of primary SS was considered.
Histopathology
Pathological testing showed protein deposits characteristic of amyloidosis in the lesions. Examination of the biopsy samples, performed using Congo red staining, was positive, and polarized light microscopy revealed characteristic apple-green birefringence (Figure 4). Furthermore, the samples showed strong expression of the kappa (κ) light chain (++) and mild expression of the Lambda (λ) light chain (+); amyloid protein expression was not noted (–) (Figure 5). The result indicated that the disease type was light-chain (AL) amyloidosis.

Histological examination of biopsy samples by using Congo red staining and polarization microscopy. (a) Congo red staining and (b) polarization microscopy.

Amyloidosis with light chain kappa expression (Kappa ++; Lambda +; AA −).
Treatment course and follow-up
Because the patient did not experience complications related to airway stenosis, the primary focus of treatment was on managing SS. The therapeutic regimen for the management of SS and hemoptysis included Iguratimod 25 mg administered twice daily (Iguratimod offers a promising targeted, oral, and well-tolerated option for SS, particularly for patients with systemic involvement.)6,7 prednisone 25 mg once daily, and hydroxychloroquine 200 mg twice daily to address.
Hemoptysis did not recur over a 6-month follow-up period. However, the amyloidosis-related lesions in the patient’s airway did not decrease in size, and additional lesions developed in the trachea and left main bronchus (Figure 6).
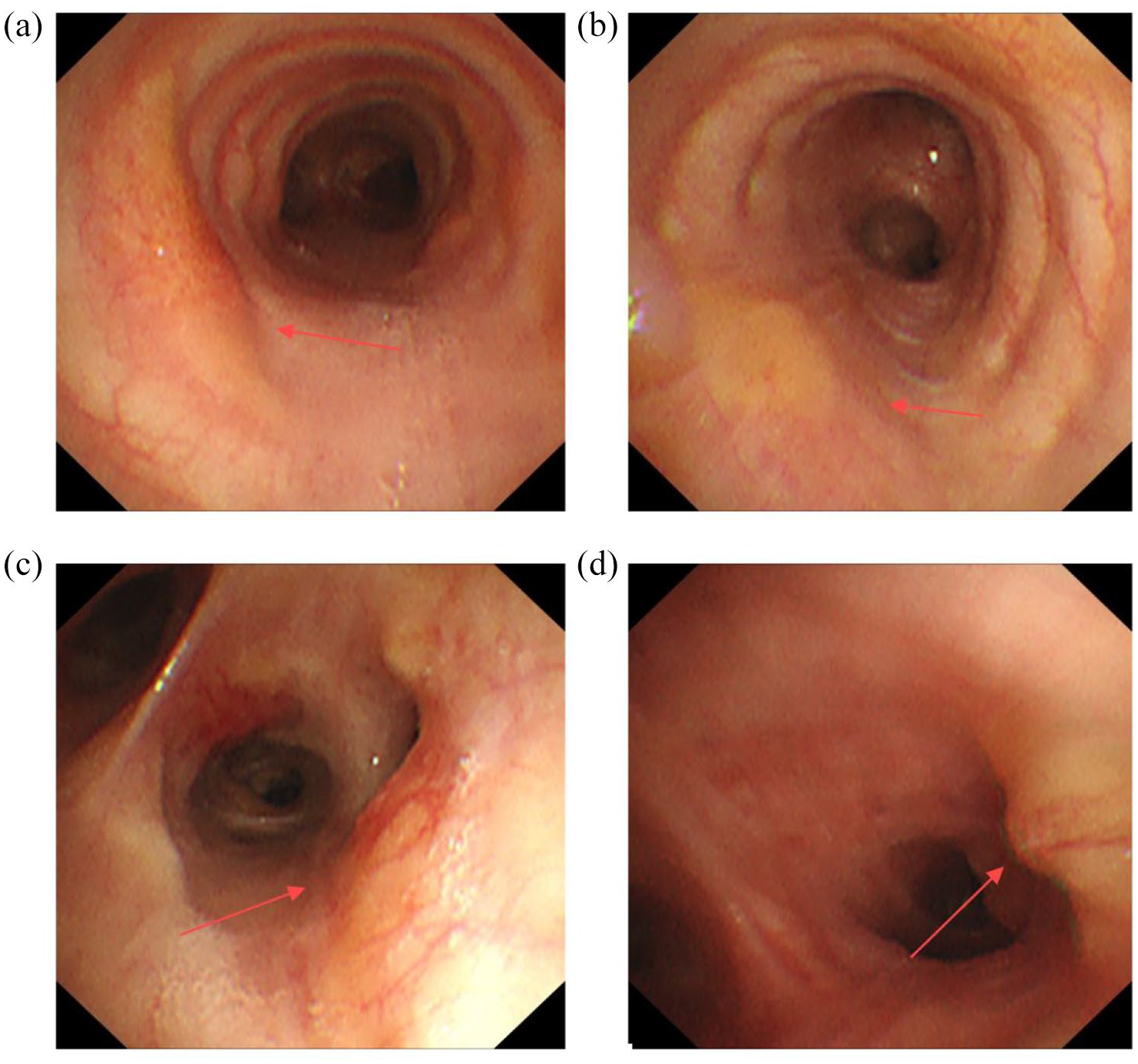
Bronchoscopy images of the patient after treatment. (a) Lesions in the trachea; (b) lesion in the left main bronchus; (c) lesions in the right intermediate bronchus; (d) lesions in the left lower bronchus.
Discussion
Here, we presented a case of TBA concomitant with SS, detailing the patient’s clinical characteristics, CT and bronchoscopy findings, relevant laboratory test results, and pathological features, with an aim to enhance understanding of this comorbidity.
Amyloidosis encompasses a spectrum of disorders characterized by the aggregation and accumulation of abnormal extracellular protein, known as amyloid, within tissues and organs, resulting in diverse clinical manifestations. Amyloid deposition can be either localized or systemic. Further, amyloidosis is classified according to the type of amyloid protein involved, with the two most prevalent forms being light-chain (AL) amyloidosis and reactive (AA) amyloidosis, the latter of which develops secondary to chronic inflammatory disease. 1 And TBA is marked by the accumulation of amyloid proteins within the airway tissues.
TBA is an exceptionally rare manifestation of SS, typically associated with localized AL amyloidosis. The underlying mechanism remains unclear, but chronic antigenic stimulation and B-cell dysregulation may promote amyloid deposition. The predominant κ light-chain expression in this patient aligns with previous reports linking SS to localized AL amyloidosis with B-cell hyperactivity.8 –13
The clinical manifestations of TBA are contingent upon the location and extent of amyloid deposits. Patients with airway amyloidosis do not exhibit specific symptoms; instead, they commonly present with cough, dyspnea, stridor, hoarseness, and hemoptysis. These symptoms are analogous to those observed in other respiratory conditions, thereby complicating early and accurate diagnosis. Amyloid accumulation in the airways results in a progressive buildup of amyloid material, leading to the thickening and rigidity of the airway walls. This, in turn, results in the narrowing of the airway lumen, contributing to respiratory symptoms and complications.
The diagnosis of TBA is typically established through a combination of clinical evaluation, imaging studies, and histopathological examination. In some patients, CT may reveal thickened airway walls and luminal narrowing. However, smaller lesions may not be detectable on CT, making bronchoscopy an essential diagnostic procedure. Bronchoscopy permits direct visualization of the airway and enables tissue biopsy. Whitish, waxy, or nodular lesions identified on bronchoscopy are indicative of amyloid deposits. Histological examination of biopsy samples by using Congo red staining in conjunction with polarization microscopy is crucial for definitive diagnosis. 2 In addition, this method facilitates the differentiation of protein components, including the κ and λ light chains, and amyloid protein.
Given the absence of systemic involvement, treatment primarily focused on managing SS-related inflammation. However, persistent airway lesions suggest a need for more aggressive interventions. While there is no standardized treatment for TBA, bronchoscopic debulking or laser therapy could be considered in cases with progressive airway obstruction.
Some studies have shown that while AA (secondary amyloidosis) amyloid deposition is more common in inflammatory diseases such as rheumatoid arthritis and ankylosing spondylitis. AL (primary) amyloidosis is mostly seen in cases of SS (Table 1). The coexistence of amyloidosis and primary SS is uncommon, with cutaneous localized amyloidosis being particularly rare. Approximately 25% of reported cases of cutaneous localized amyloidosis are associated with SS. 14 Amyloid involvement of lungs is very rare in SS and occurs in 0%–2% of symptomatic patients with pulmonary infiltrates.13,15 –19 We have presented a case of TBA in a patient with SS. Other rare respiratory complications, such as lymphoma and pulmonary hypertension, must be considered in patients with SS.3,4
Previous studies about coexistence of Sjögren’s syndrome and tracheobronchial amyloidosis.

NA, not applicable.
Conclusion
This case underscores the importance of considering TBA in Sjögren’s syndrome patients presenting with unexplained respiratory symptoms. The diagnosis requires a high index of suspicion, supported by CT imaging, bronchoscopy, and histopathological confirmation. Although systemic immunosuppression may control underlying autoimmune activity, localized amyloid deposition often persists, necessitating multidisciplinary management with potential bronchoscopic intervention. Further research is needed to establish optimal treatment strategies for this rare and challenging condition.
Supplemental Material
sj-docx-1-tar-10.1177_17534666251342145 – Supplemental material for Tracheobronchial amyloidosis with Sjögren’s syndrome: a case report
Supplemental material, sj-docx-1-tar-10.1177_17534666251342145 for Tracheobronchial amyloidosis with Sjögren’s syndrome: a case report by Chongxiang Chen, Pingping Wang, Na Zhao, Derong Zhang, Cuifen Chen and Ping Peng in Therapeutic Advances in Respiratory Disease
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.