
Abstract
Acute exacerbation of chronic obstructive pulmonary disease (AECOPD) is the main cause of hospitalization and death of patients with chronic obstructive pulmonary disease. This is largely due to bacterial resistance caused by clinical antibiotic abuse and the limited efficacy of current treatment strategies in managing noninfectious AECOPD, which presents a significant challenge for clinicians. Therefore, it is urgent for clinical treatment and prevention of AECOPD to fully understand the specific mechanism of AECOPD in the immune system and master the key differences between infectious factors and noninfectious factors. This article systematically discusses AECOPD triggered by various factors, including the activation of immune system, the recruitment and activation of inflammatory cells and the role of specific inflammatory responses, and through a comprehensive review of the literature, this article expounds the existing targeted diagnosis and treatment methods and technologies at different stages in order to provide new ideas and strategies for clinical prevention and treatment of AECOPD.
Plain language summary
Chronic obstructive pulmonary disease (COPD) can get worse suddenly, leading to what doctors call an acute exacerbation (AECOPD). These flare-ups are the main reason why people with COPD end up in the hospital or die from the disease. One problem is that overusing antibiotics can make infections harder to treat, and noninfectious causes of AECOPD are often not well treated. To better treat and prevent AECOPD, it’s important to understand exactly how these flare-ups affect the immune system and to know the key differences between those caused by infections and those that aren’t. This paper explains the different reasons why AECOPD happens, including how the immune system is activated, how inflammatory cells are involved, and the specific inflammatory reactions that occur. We also review current methods and technologies for diagnosing and treating AECOPD at different stages. This information aims to provide new ideas and strategies for preventing and treating AECOPD in clinical practice.
Introduction
Chronic obstructive pulmonary disease (COPD) remains a significant global health challenge, ranking as the third leading cause of mortality. 1 The acute exacerbations (AE) of COPD are the primary drivers of hospitalization and mortality among these patients. 2 AECOPD, a clinical condition characterized by rapid deterioration of airway function and respiratory symptoms, affects a substantial proportion of COPD patients, with 22%–40% experiencing at least one exacerbation per year and 9%–16% experiencing multiple attacks. 3 The triggering factors for AECOPD encompass both infectious and noninfectious components. Infectious factors typically refer to infections caused by pathogens such as bacteria, viruses, and fungi, which subsequently activate the immune system and provoke inflammatory responses. In contrast, noninfectious factors include environmental elements (such as air pollution and smoke), allergic reactions, medications, and psychological stressors, all of which can lead to airway inflammation and dysfunction. 4 Notably, bacterial infection accounts for approximately 50% of AECOPD cases. 5 Antibiotic treatment forms a crucial component of conventional therapy for AECOPD. 6 However, the therapeutic efficacy is often limited by the presence of noninfectious factors and antimicrobial resistance, leading to unsatisfactory outcomes and significant economic burden in clinical practice. 7
Therefore, it is important to understand the different changes of immune system in vivo to infectious factors and noninfectious factors. 8 Only in this way can we better decide the use of antibiotics, improve the curative effect of patients, reduce the economic burden of patients, and improve the quality of life. Therefore, this review will begin by examining both infectious and noninfectious factors, detailing the mechanisms underlying the patients’ immune responses, as well as discussing the latest research and corresponding treatment methods. This aims to provide new insights for the prevention and treatment of clinical AECOPD.
Activation of the immune system
COPD is a chronic inflammatory lung disease characterized primarily by airflow limitation, which is usually not fully reversible. This condition arises because various stimuli (such as smoking, harmful particles or gases, pathogens) activate airway epithelial cells and pulmonary macrophages, leading to the release of multiple inflammatory mediators (such as IL-8 and TNF-α). These mediators attract inflammatory cells, including neutrophils, macrophages, and lymphocytes, to the airways, causing an inflammatory response in the lungs. Prolonged inflammation results in thickening, fibrosis, and hyperplasia of the airway walls, leading to narrowing of small airways and bronchial wall destruction, ultimately causing airflow limitation. Furthermore, the destruction of alveolar walls leads to emphysema, which further reduces the surface area for gas exchange. 9 This inflammatory basis underlies the physiological propensity for AECOPD when exposed to various stimuli. Understanding the mechanisms by which different factors lead to AECOPD is essential for improving patient outcomes (Figure 1).
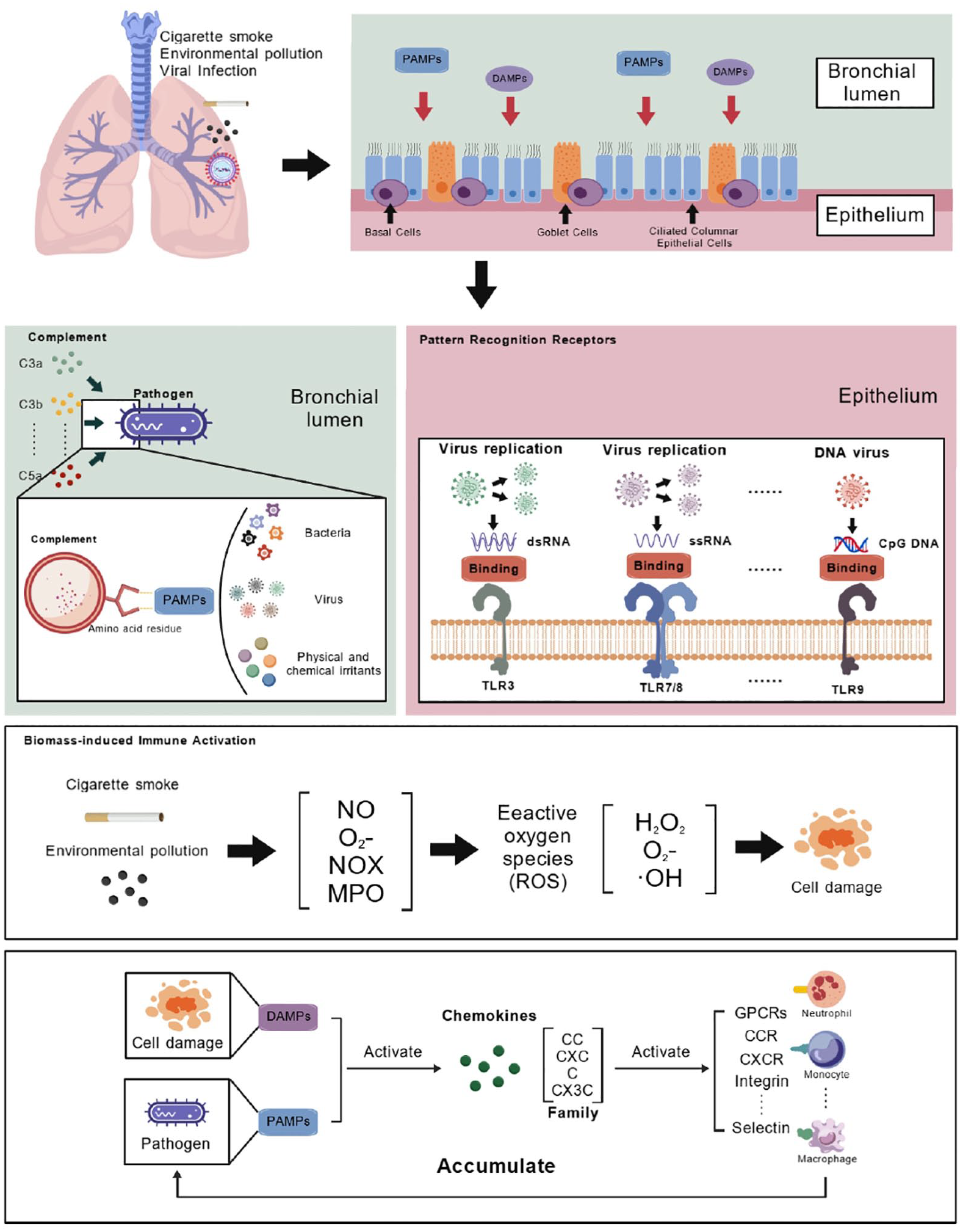
Schematic diagram of infectious factors and noninfectious factors invading epithelial cells of COPD patients.
Infection-driven immune activation
Free pathogens stage
Pathogens enter the respiratory tract through inhalation or contact and are recognized with the help of airway epithelial cells, macrophages, and dendritic cells. Pattern recognition receptors (PRRs) on host cell surfaces (such as TLRs and NLRs) identify pathogen-associated molecular patterns (PAMPs), initiating innate immune responses. Common pathogens such as bacteria, fungi, and viruses can enter the lungs via the respiratory tract. Bacteria, for example, adhere to respiratory epithelial cells using surface adhesins (such as capsules, polysaccharides, proteins), primarily infecting areas like the nasal cavity, pharynx (e.g., Group A Streptococcus causing pharyngitis), trachea, bronchi, and alveoli (e.g., Streptococcus pneumoniae causing pneumonia, Klebsiella pneumoniae). 10 Fungi spread through airborne spores, affecting areas such as the sinuses (e.g., Aspergillus causing sinusitis) and bronchi and alveoli (e.g., Aspergillus and Candida causing pulmonary mycosis). 11 Viruses attach to respiratory epithelial cells through specific surface receptors (e.g., hemagglutinin of influenza virus binding to sialic acid receptors), affecting regions such as the nasal cavity, pharynx (e.g., rhinovirus causing the common cold), and bronchi and alveoli (e.g., influenza and SARS-CoV-2 causing pneumonia). 12
Upon entry into the body of COPD patients, the complement system plays a crucial role in the initial stages of infection. 13 Opsonization by complement is highly effective in clearing bacteria, viral particles, and fungi. The complement system is activated via classical, alternative, or lectin pathways, 14 producing active fragments like C3b during the complement cascade. C3b is the primary component of opsonization, covalently binding to pathogen surfaces (such as bacteria and viral particles), marking these pathogens for recognition. These opsonized pathogens interact with complement receptors (such as CR1) on phagocytes (e.g., macrophages and neutrophils), enhancing phagocyte affinity for the pathogens and promoting their engulfment and destruction. 15 Clinically, lower C3 levels in AECOPD patients are associated with poorer prognosis. 16 Beyond opsonization, noninactivated C3b on cell surfaces combines with factor B to form C3 convertase (C3bBb), leading to more C3 activation and formation of C5 convertase (C3bnBb). This process results in the formation of the membrane attack complex, which punctures pathogen membranes, causing lysis and death. 17
It’s important to note that the complement system has precise regulatory mechanisms. For instance, C3b deposited on cell surfaces is rapidly inactivated by regulatory proteins unless it is on surfaces lacking these proteins (e.g., pathogen surfaces), where it remains active. 17 Thus, under normal circumstances, the complement system does not damage normal cells. At present, the clinical use of the C5 complement specific drug Eculizumab (Soliris®, Alexion Pharmaceuticals, Boston, MA, USA) has been approved. It is a new human igG2/4k monoclonal antibody, which was produced by NS0 cell line using recombinant DNA technology. It binds to C5 to prevent C5 invertase from cracking C5a and C5b-9 complex. Eculizumab is approved for the treatment of paroxysmal nocturnal hemoglobinuria. However, in a placebo-controlled trial, the drug inhibited C5 in the lung by inhalation and showed significant response in improving vital capacity and reducing eosinophilia induced by allergens in the middle and late stage of induced sputum in patients with mild allergic asthma. 18 Then it also has a certain prospect in the treatment of COPD patients.
Furthermore, enhancing complement system activity in AECOPD patients to suppress pathogen invasion is crucial. Researchers are exploring macrophage-avoiding nanoparticles (NPs) designed with Poly(2-methyl-2-oxazoline) (PMOXA), which have been shown in animal models to broadly trigger complement activation via the classical pathway and accelerate C3 opsonization. 19
Pathogen–host cell interaction
PRRs are widely expressed in cells involved in innate immune responses. They recognize PAMPs and Damage-Associated Molecular Patterns (DAMPs), playing a crucial role in host defense by detecting foreign pathogens or endogenous damage signals and initiating appropriate immune responses. 20
Upon contact with normal tissue cells of COPD patients, pathogens are first recognized by host cell PRRs. Key PRRs include Toll-like receptors (TLRs), Nod-like receptors (NLRs), Rig-like receptors, scavenger receptors (SRs), and cytoplasmic DNA sensors. TLRs, in particular, play a significant role in AECOPD due to their broad recognition capabilities and inflammation-regulating functions.21,22 Eleven types of TLRs have been identified in humans, each recognizing different PAMP components and molecules. For example, double-stranded RNA (dsRNA) produced by many viruses during replication is recognized by TLR3; single-stranded RNA (ssRNA) viruses such as influenza virus and HIV are recognized by TLR7 and TLR8; unmethylated CpG DNA sequences in DNA viruses are recognized by TLR9.23 –25 Upon activation, TLRs initiate different signaling pathways. Clinically, TLR4 expression in bronchial epithelial cells positively correlates with COPD severity. 26 Further research shows that lipopolysaccharide (LPS), a common PAMP, is recognized by TLR4 on host cells. LPS binding to TLR4, facilitated by the MD-2 auxiliary protein, promotes TLR4 dimerization and activation. Activated TLR4 then binds MyD88 adaptor protein via its TIR domain, recruiting and activating IL-1 receptor-associated kinases (IRAKs), which subsequently activate TNF receptor-associated factor 6 (TRAF6). TRAF6 mediates IκB kinase (IKK) activation, leading to IκBα phosphorylation and degradation. Degraded IκBα releases NF-κB dimers (e.g., p65/p50), which translocate to the nucleus to initiate target gene transcription, inducing the expression of TNF-α, IL-1β, IL-6, MCP-1, and other inflammatory mediators, thereby exacerbating local and systemic inflammatory responses.27 –32 In addition to TLR4’s importance in COPD progression, Pseudomonas aeruginosa flagellin stimulates TLR5, leading to phosphorylation of p38, ERK, and JNK, which triggers the production of IL-6 and IL-8, promoting COPD progression and exacerbation. 33 Research using lung tissue explants infected with viruses shows that activation of TLRs 3, 7, and 8 in lung tissue stimulates the release of pro-inflammatory molecules like TNFα and CCL5, triggering inflammatory responses. 34 Thus, TLRs play a significant role in the occurrence and development of AECOPD.
Current clinical applications of TLRs in AECOPD focus on reducing tissue damage and enhancing antiviral capabilities. Inhibitors of the TLR4 signaling pathway, such as TAK-242 and Bay 117082, have been developed, significantly inhibiting inflammatory responses in pulmonary artery smooth muscle cells of COPD rats and reducing IFN-γ production.35,36 The PDE4 inhibitor piclamilast modulates TLR7/8 signaling pathways to inhibit inflammation caused by Rhinovirus. 37 Additionally, TLR5 receptors play an important role in enhancing mucosal immune responses, protecting the respiratory tract from pathogens, and increasing resistance to bacterial infections, proving beneficial in COPD treatment. 38
Biomass-induced immune activation
Exacerbations in COPD patients can also be triggered by various noninfectious factors, including environmental pollution (particulate matter and toxic gases such as PM2.5, sulfur dioxide, and nitrogen oxides), smoking, climate changes (cold weather, humidity variations), improper medication use, and the exacerbation of comorbidities (such as cardiovascular diseases). 39 Noninfectious factors significantly contribute to AECOPD incidence and remain a major clinical challenge.
Smoking is a predominant noninfectious factor and a leading cause of COPD. Smoking and air pollution generate large amounts of reactive oxygen species (ROS), leading to lung cell integrity damage and inflammatory response activation. 40 ROS are reactive molecules and free radicals containing oxygen, including hydrogen peroxide (H2O2), superoxide anion (O2–), and hydroxyl radical (·OH). While ROS are normally generated during cellular metabolism, excessive ROS cause cell damage and inflammation. Smoking and air pollution generate ROS through various mechanisms, such as the direct production of ROS from nitric oxide (NO) and superoxide (O2–) in cigarette smoke and the activation of lung neutrophils and macrophages, producing large amounts of ROS through NADPH oxidase (NOX) and myeloperoxidase (MPO).40 –42 Excessive ROS directly oxidize cellular lipids, proteins, and DNA, leading to cell dysfunction and death. Importantly, ROS induce lipid peroxidation, protein carbonylation, and DNA histone modifications, 43 which act as DAMPs, recognized by PRRs, activating the innate immune system.
Unlike TLRs that predominantly recognize PAMPs, NLRs play a major role in AECOPD caused by noninfectious factors. The NLR family has 22 members in humans and more in mice, most sharing common structural features, including a C-terminal leucine-rich repeat domain involved in ligand recognition, a central nucleotide-binding oligomerization domain (NOD), and a variable N-terminal effector domain. 44 NLRs are further classified into five subfamilies. The NLRA subfamily includes CIITA, a transcription factor with at least one splice variant expressing CARD. CIITA activates transcription of genes encoding major histocompatibility complex class II. 45 The NLRB group includes NAIP1-7 in mice and NAIP in humans, expressing a BIR domain. The NLRC group includes NOD1 and NOD2, expressing CARD. The NLRP group includes NLRP1-14, expressing PYD. The NLRX group contains only one member, NLRX1, expressing CARD, and localized to mitochondria.44,46 Among these, NLRP3 plays a critical role in noninfectious AECOPD, responding to a variety of DAMPs, including ROS. Studies have shown that components in cigarette smoke can induce stress responses and damage in airway epithelial cells and macrophages, leading to the release of cytokines and chemokines that activate NOD1 and NOD2. Activated NOD1 and NOD2 then trigger downstream signaling pathways (such as NF-κB and MAPK), further promoting the release of inflammatory mediators and exacerbating airway inflammation and tissue damage. 47 Moreover, smoking increases ROS production in mitochondria, which activates NLRX1, resulting in mitochondrial dysfunction. 48 Dysfunctional mitochondria also produce excessive ROS, leading to persistent immune system stimulation. 49
Researchers have found that Nlrp10 gene knockout mice exposed to cigarette extract exhibited significantly lower leukocyte recruitment compared to wild-type mice. Under the influence of cigarette extract, the expression of Nlrp10 protein not only promotes leukocyte aggregation but also plays a crucial role in caspase-1 activation, cytokine/chemokine production (IL-1β, IL-18, MCP-1, and IL-17A), and the induction of NF-κB and MAPKs in the lungs of C57Bl/6 mice. 50 Cell experiments have also shown that exposure to cigarette extract increases the production of inflammatory cytokines (IL-8 and MCP-1) in a dose-dependent manner. Additionally, cells exposed to both cigarette extract and LPS exhibit significantly increased NLRP3 inflammasome activity and IL-1β levels. 51
Extensive clinical and experimental studies have confirmed the importance of NLR in the progression of COPD due to noninfectious factors such as smoking. Consequently, research into treating AECOPD by inhibiting the function of NLR receptors and related signaling pathways has emerged. Isoforskolin and sodium butyrate appear promising. Researchers have found that Isoforskolin can alleviate AECOPD by downregulating pro-inflammatory cytokines, Th17/IL-17A, and the NF-κB/NLRP3 pathway, thereby improving lung function and reducing inflammation. 52 Sodium butyrate mitigates smoking-induced COPD inflammation by activating GPR43 and inhibiting the NF-κB/MAPKs signaling pathway. It also inhibits the expression levels of the NLRP3 inflammasome, which comprises NLRP3, apoptosis-associated speck-like protein (ASC), and caspase-1 in cells stimulated by cigarette extract. 53 Additionally, other drugs such as MCC950, a potential treatment for NLRP3-related syndromes (including autoinflammatory and autoimmune diseases), may also become potential treatments for COPD. 54
Inflammatory cell recruitment and activation
Selectins
In COPD, immune system activation due to multiple pathways leads to elevated levels of various inflammatory mediators in the airway, lung tissue, and bloodstream. 55 The continuous migration of inflammatory cells (mainly neutrophils) from the vascular compartment to the lungs is partially regulated by selectins. 56 Selectins mediate transient adhesive interactions related to inflammation by recognizing carbohydrate epitopes such as sialyl Lewisx (sLex) on structurally diverse protein–lipid ligands expressed on circulating leukocytes. The rapid turnover of selectin-ligand bonds, due to their fast off-rates and very high tensile strength, enables them to mediate cell tethering and rolling in shear flow. 57 Three selectins have been identified: L-, P-, and E-selectins. Targeting these molecules may reduce inflammation in COPD. 58 Inhibiting selectin function is commonly achieved by directly inhibiting one or more selectins. Carbohydrates, recombinant soluble ligands, antibodies, and small molecule inhibitors have all entered clinical development as potential therapeutics targeting selectins.
Heparin is a known inhibitor of selectin-mediated interactions. PGX-100 (2-O, 3-O desulfated heparin) and PGX-200 (the inhaled formulation of PGX-100) have been developed to maximize heparin’s anti-selectin activity while minimizing anticoagulant effects, but PGX-100’s phase IIb trial in AECOPD patients was terminated due to lack of efficacy shown in interim analysis. 59 Bimosiamose (TBC1269,1) is a synthetic pan-selectin antagonist targeting E-, P-, and L-selectins. In vitro, under static and dynamic flow conditions, bimosiamose blocks adhesion of neutrophils, eosinophils, and lymphocytes. 60 In vivo, it has shown anti-inflammatory effects in various animal models, including lung inflammation. 61 In a pilot trial, inhaled bimosiamose was safe and well-tolerated in stable mild-to-moderate COPD patients and showed positive anti-inflammatory effects on sputum parameters, reducing IL-8 levels and lymphocyte counts. 62 Additionally, a study of 77 moderate-to-severe COPD patients using standard bronchodilators showed that 28 days of inhaled bimosiamose was well-tolerated and safe, 63 leading to widespread and significant attenuation of airway inflammation and trends toward improved lung function.
Chemokines
Various chemokines are major drivers of local inflammatory cell aggregation. Chemokines are small molecules (8–12 kDa) belonging to the cytokine family. Due to their involvement in various biological functions such as chemotaxis, leukocyte degranulation, hematopoiesis, and angiogenesis, they are therapeutic targets in many inflammation-related diseases.64 –67 Chemokines mediate various cellular processes, particularly chemotaxis, through interactions with cell surface G-protein-coupled receptors. More than 50 different chemokines are known to activate up to 20 different surface receptors. 68 Based on differences in the position of key cysteine residues, chemokines are classified into four families: CC, CXC, C, and CX3C. These four groups of chemokines are defined by the positions of conserved residues and their quaternary structures. 69 All chemokines share a highly conserved tertiary structure fold, consisting of a flexible N-terminus and N-terminal loop, followed by a three-stranded β-sheet with a C-terminal α-helix. This conformation allows them to form oligomers, mainly not only homodimers and heterodimers but also tetramers and higher-order oligomers. 70
Chemokines strongly bind to glycosaminoglycans (GAGs), and this interaction drives chemokine oligomerization. 71 Oligomerized chemokines can form more stable and defined chemical gradients in tissues, guiding inflammatory cells along the gradient to specific sites of inflammation. 72 For instance, CXCL8 or IL-8 binds to heparan sulfate, promoting the stabilization and distribution of chemokines in the extracellular matrix, forming chemical gradients that attract neutrophils and other inflammatory cells to sites of infection or injury. Different oligomers, such as monomers and dimers of CXCL12, selectively induce different signaling pathways, such as β-arrestin recruitment, with only monomeric CXCL12 promoting cell migration.73,74 For some chemokines, such as CCL2, CCL4, and CCL5, GAG binding and oligomerization are necessary to induce cell recruitment. 75 In the context of AECOPD, chemokine gradients in lung tissue differ significantly from those in other locations, mainly due to lung extracellular matrix (ECM) components, especially GAGs. 76 ECM proteins have been shown to further influence chemokine expression levels and localization patterns during AECOPD.77,78
Among many chemokines, CXCL8 (IL-8) is one of the most extensively studied and well-known neutrophil inflammatory mediators.79 –81 Initially identified as a monocyte-secreted factor stimulated by lipopolysaccharide (LPS) that stimulates neutrophil degranulation and oxidative burst, 82 CXCL8 was the first chemokine found to play a role in COPD inflammation. Compared to smokers with normal lung function and nonsmokers, COPD patients show elevated CXCL8 concentrations in induced sputum, correlating with increased neutrophil numbers. 79 Subsequent studies have confirmed CXCL8’s role in neutrophil inflammation in COPD airways.
Other chemokines also play significant roles in COPD progression. CXCL9, CXCL10, and CXCL11 are primarily produced by alveolar macrophages but can also be produced by bronchial epithelial and bronchial smooth muscle cells.50,83 –86 Their affinity for their common receptor (CXCR3) increases from CXCL9 to CXCL11, indicating a hierarchical relationship. 87 Elevated concentrations of these chemokines in induced sputum from COPD patients correlate with neutrophil numbers. 88 COPD patients’ peripheral blood mononuclear cells exhibit enhanced migratory responses to CXCL9, CXCL10, and CXCL11 compared to nonsmokers. 89 Activated Th1 and CD8+ lymphocytes induce CXCR3 expression and are believed to be involved in recruiting these cells to inflammation sites in COPD patients. 90
Further research has confirmed that the decline in lung function in COPD patients is associated with a higher proportion of CXCR3-expressing T cells, 91 suggesting that CXCL9, CXCL10, and CXCL11 may be involved in T cell recruitment and subsequent immune-mediated lung injury observed in COPD. 92 Conversely, the CXCL11 gradient in the lungs of COPD patients is also associated with the resolution of lung inflammation. The recruitment of effector T cells to mucosal surfaces, such as the bronchial epithelium, is important for defending against airway pathogens, but the migration of lymphocytes from the epithelium to the airway lumen, known as “egress,” is also physiologically crucial for preventing harmful leukocyte accumulation in the stroma.
Inflammatory response
Upon migration of immune cells such as neutrophils, monocytes/macrophages, and T lymphocytes to the lung tissue through chemokines, these cells release additional inflammatory mediators locally, thereby exacerbating the local inflammatory response.
Neutrophil recruitment and activation
Neutrophils are a critical component of the host response against invading pathogens, representing the first line of defense in the innate immune system, accounting for approximately 70% of peripheral blood leukocytes. 93 In response to inflammatory stimuli, neutrophils migrate from the circulation to the site of infection, where they can efficiently bind, phagocytize, and inactivate pathogens through phagocytosis, degranulation, and the release of neutrophil extracellular traps (NETs). 94 When the body is infected or stimulated to induce inflammation, neutrophils become activated and release NETs, composed of granules and nuclear components, which can neutralize and kill pathogens extracellularly through a series of activated signaling pathways. However, the impact of NETs can be potentially harmful depending on the location, timing, and degree of the inflammatory response. Excessive production of NETs at specific times or locations can lead to tissue damage in the host.
One significant factor contributing to the development of AECOPD is excessive NETs damage. Studies have found the presence of NETs in the airways of patients with chronic inflammatory airway diseases. 95 Furthermore, the accumulation of NETs is associated with the activation of innate immune responses, which may help to understand the pathogenesis of AECOPD induced by neutrophils. 95 Various biological molecules produced by pathogens or biologics, such as interleukin-8 (IL-8), tumor necrosis factor-alpha (TNF-α), platelet-activating factor, lipopolysaccharides (LPS), anti-neutrophil cytoplasmic antibodies, granulocyte-macrophage colony-stimulating factor (GM-CSF), and complement factor 5a (C5a), have been shown to induce neutrophil activation.96 –101 Subsequently, activated nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX2) produces large amounts of ROS, leading to neutrophil nuclear membrane rupture through interaction with Toll-like receptor 4 (TLR4). Moreover, intracellular neutrophil elastase (NE) and myeloperoxidase (MPO) migrate to the nucleus through the ruptured neutrophil nuclear membrane, where they partially degrade specific histones and promote chromatin decondensation. 102 Additionally, histone citrullination induced by peptidyl arginine deiminase 4 (PAD4) further promotes chromatin decondensation. 103 The exact mechanism of PAD4 activation remains largely unknown, although one study suggests that calcium binding may be involved, and ROS may play a role in regulating PAD4 activation. 104 After chromatin decondensation, the loose chromatin mixes with granular cytoplasmic and various proteins, which are then extruded into the extracellular space, forming a web-like structure.
The release of NETs triggers a form of cell death known as NETosis, distinct from apoptosis and necrosis. 105 NETosis is an irreversible process that is highly dependent on ROS production, referred to as NOX-dependent NETosis. However, a groundbreaking report demonstrated that during acute infection with Staphylococcus aureus, neutrophils retained multitasking abilities upon releasing NETs, found to be independent of NOX. 106 Current research reports indicate that platelet TLR4 and activated PAD4 play crucial roles in NETs formation,98,107 and some studies have shown that direct interaction between neutrophils and LPS, without involving platelet TLR4, can also lead to NET formation.108 –110 The mechanisms of NETosis are still not fully understood. However, it is well known that NOX activation plays an important role in NETosis, with NOX-dependent NETosis signaling cascades involving the Raf/MEK/ERK and p38 MAPK pathways. 111 Additionally, the TLR4-MyD88 signaling pathway is necessary for neutrophils to release NETs.112,113
Neutrophils can induce chronic airway mucus hypersecretion and lung parenchymal destruction by releasing NE, a major component of NETs and other active substances. NE has a pro-inflammatory effect in COPD, especially by stimulating the secretion of IL-8. 114 Therefore, COPD is a significant influencer of tissue damage mediated by NETs formation and NETosis. Clear morphological evidence indicates the presence of NETs in the sputum of stable and exacerbated COPD patients.115,116 Furthermore, a study by Grabcanovic-Musija et al. showed that NETs formation in COPD patients correlates with the severity of airflow limitation. 116 The exact pathophysiological mechanisms of NETs involvement in airway inflammation and lung injury in COPD patients remain unclear. NETs, containing a mixture of extracellular DNA, histones, and granular proteins, may directly exert cytotoxic effects on airway epithelial and endothelial cells or indirectly induce lung tissue damage by promoting autoimmune responses against abnormal amounts of NET components.117,118 The precise mechanisms remain unknown.
In noninfectious COPD progression, the relationship with neutrophils appears to be minimal. Some studies suggest that nicotine toxicity-induced lung injury can occur in smokers without manifesting airflow limitation, as nicotine, the addictive component of tobacco, may cause nicotine toxicity.116,119 Additionally, studies have shown that the current smoking status of COPD patients does not affect NET formation, 120 indicating that smoking may not trigger NET formation in COPD patients. Currently, glucocorticoids are the primary treatment for AECOPD. While some patients with AECOPD respond favorably to systemic glucocorticoid treatment, a subset of patients remains resistant to this therapy. 121
Adaptive immunity
Mature T lymphocytes exhibit significant heterogeneity, characterized by differences in cell membrane molecule expression and biological functions. According to the type of T cell receptor (TCR) molecules, they can be divided into αβ T cells and γδ T cells, with the former being the majority and serving as the primary effector cells mediating adaptive immune responses. αβ T cells can also be classified into helper T lymphocytes (Th), cytotoxic T lymphocytes (Tc), and regulatory T lymphocytes (Treg) based on functional characteristics. Th cells mainly secrete cytokines to promote the activation and function of other cell types, exhibiting extensive immunoregulatory effects. Tc cells, known for their direct cytotoxic effects, play an important immunoprotective role against viral infections. Treg cells, named for their broad immunosuppressive/regulatory functions, play a crucial role in maintaining self-tolerance, peripheral tolerance, and inhibiting pathological immune damage.
Studies have shown that, compared to asymptomatic smokers with normal lung function, the number of CD8+ T lymphocytes is increased in the lung tissue of COPD smokers. 122 Additionally, the content of CD8+ T cells in the sputum of COPD patients is also elevated. 123 This suggests a potential correlation between the severity of COPD and the levels of CD8+ T cells, and to explore the relationship between CD8+ T cells and COPD disease, researchers used cell sorting techniques to find that the abundance of two CD8+ T cell subsets increased in the lungs of mild-to-moderate COPD patients: cytotoxic klrg1+TIGIT+CX3CR1+TEMRA (T effector memory CD45RA+) cells and DNAM-1+CCR5+ T resident memory (TRM) cells. These CD8+ T cells interact with myeloid pulmonary type II cells through Interferon gamma (IFNG) and have overly expanded TCR clonotypes, potentially leading to inflammation before severe disease. 124 Interestingly, these T cell subsets exhibit different spatial distributions in COPD patients, revealing their specific pathological features (e.g., alveolar destruction, airway metaplasia) driven in COPD. TEMRA cells, primarily found in the blood and lymph nodes, 125 reflect pulmonary circulation rather than permanent residency in the lungs. Imaging mass spectrometry technology shows immune subgroups around the terminal bronchioles in COPD, significantly associated with small airway remodeling and the presence of infiltrative CD8+ T cells. After the initial infection, selective CD8+ T cells establish residency in the lungs (TRM), maintaining immune memory to respond to future challenges from the same pathogen.
This immune cell residency at the terminal bronchioles significantly impacts COPD patients’ condition. COPD is characterized by extensive immune cell infiltration and structural changes such as peribronchial fibrosis. Fibrocytes, derived from the bone marrow and released into the peripheral circulation, are associated with pulmonary fibrosis. 126 They are recruited into the blood of COPD patients during AE. 127 High circulating fibrocyte counts during this stage correlate with increased mortality risk, suggesting that fibrocytes may be detrimental to disease progression. 127 Besides their role in scar matrix generation and affecting pulmonary contraction, recruited fibrocytes may participate in lung inflammation through their immune properties, acting as antigen-presenting cells for T cells, thus regulating fibrocyte differentiation. 128
Researchers have found that fibrocytes and CD8+ T cells are located near the distal airways. Compared to the control group, the potential interactions between these two cell types are more frequent in COPD patient tissues. This increased proximity and clustering of CD8+ T cells and fibrocytes correlate with changes in patients’ lung function. CD8+ T cells from COPD patient tissues promote fibrocyte chemotaxis via the CXCL8-CXCR1/2 axis. Live imaging shows that CD8+ T cells establish short-term interactions with fibrocytes, triggering CD8+ T cell proliferation, pro-inflammatory cytokine production, cytotoxic activity of CD8+ T cells against bronchial epithelial cells, and the immunoregulatory properties of fibrocytes through a CD54- and CD86-dependent manner. 129 This offers an explanation for why infections exacerbate the condition in COPD patients.
However, not all COPD patients infected with pathogens experience exacerbations. Studies have found that the gene HHIP (hedgehog-interacting protein), which is associated with COPD susceptibility, is highly expressed in lung tissue. Haploinsufficiency of HHIP induces T cell accumulation in the lungs. 130 Additionally, fibroblast-specific deletion of HHIP enhances the accumulation of IFNγ+ tissue-resident T cells following respiratory viral infections. 131 This suggests that genetic variations driven by common mutations can alter the host’s susceptibility to CD8+ T cell residency, providing another explanation for why some clinical COPD patients develop AECOPD following infections.
Smoking also affects COPD patients’ susceptibility to viral infections. Cigarette smoke (CS) induces chronic pulmonary inflammation and is a key etiological factor in the development and progression of COPD. Long-term exposure to inhaled irritants activates structural cells and inflammatory cells in the airways. 132 Studies have found that smoking impairs the antiviral CD8+ T cell immune response in COPD patients. CS interferes with the production and presentation of Major Histocompatibility Complex (MHC) class I antigens, impairing CD8+ T cell activation during viral infections. 133 Structural and inflammatory cells in the lungs respond to CS exposure by releasing pro-inflammatory mediators, which recruit additional inflammatory immune cells, collectively establishing a chronic inflammatory microenvironment. Chronic inflammation leads to lung injury, impairs innate and adaptive immune responses, and promotes recurrent respiratory infections, thereby exacerbating and further contributing to the pathological manifestations of stable disease. 134 Overall, regulating the function of immune cells like CD8+ T cells is crucial in preventing AECOPD.
Tregs play a crucial role in maintaining tolerance to self-antigens and mitigating autoimmune inflammatory responses. They are a subset of CD4+ T cells, and a reduction in Tregs can lead to excessive inflammation. A clinical cross-sectional study has identified immune phenotypes in patients with AECOPD, 135 highlighting abnormal activation of Tregs. Furthermore, patients with elevated Treg levels demonstrate significantly better lung function compared to those with low Treg levels. 136
Notably, the function of Tregs is severely impacted by smoking. Interestingly, both in the serum of COPD patients and in the bronchial epithelial (HBE) cells of individuals exposed to cigarette smoke extract (CSE), the secretion of secreted frizzled-related protein 2 (sFRP2) is significantly upregulated. Silencing the sFRP2 gene leads to a notable increase in the percentage of Tregs and a decrease in the levels of IL-6 and TNF-α in HBE cells, thereby improving the inflammatory response. 137 Additionally, miR-29b can regulate the Th17/Treg imbalance induced by CSE in experimental COPD by inhibiting the IL-22-dependent JAK/STAT3 pathway. 138 These findings provide new insights for the treatment and prevention of AECOPD caused by noninfectious factors.
Moreover, the immunometabolism of Tregs may play a significant role in the pathogenesis of COPD. For instance, glycolysis is crucial for the induction and suppressive functions of Tregs in both humans and mice, as the glycolytic enzyme enolase-1 regulates the expression of specific FoxP3 splice variants in human Tregs. 139 Investigating the immunometabolism of Tregs could offer novel therapeutic avenues for COPD. Additionally, leptin, an effective regulator of intracellular glucose metabolism and glycolysis, is a key circulating factor that increases cellular glucose uptake, in conjunction with insulin. Plasma leptin levels are significantly negatively correlated with lung function in COPD patients. 140 Understanding whether leptin can modulate FoxP3 splicing through glycolysis to influence Treg function and subsequently affect lung function in COPD patients is an important research direction.
Humoral immunity
The cells responsible for humoral immunity are B lymphocytes (B cells), also known as bone marrow-dependent lymphocytes. When viral particles or bacterial surface antigens invade, antigen-specific B cells are activated, proliferate, and ultimately differentiate into plasma cells. These plasma cells produce specific soluble immunoglobulins (Igs) or antibodies. These antibodies mediate specific immune responses by binding to and neutralizing antigens, thereby fulfilling the role of immune effectors. 141
Researchers have observed elevated levels of B lymphocytes in both central and peripheral airways of patients with COPD. 142 Furthermore, the number of B cells and B cell-rich lymphoid follicles (LFs) around small airways increases with the severity of the disease.142,143 LFs play a crucial role in the development and activation of B cells, as well as in antibody production and immune memory. In LFs, B cell-activating factor (BAFF) is produced by plasma cells and dendritic cells, significantly influencing B cell maturation, differentiation, and survival. Increased expression of BAFF has also been noted in the lungs of COPD patients and mice exposed to CS.144,145 Treatment with BAFF receptor (BAFFR)-Fc in CS-exposed mice significantly reduces the number of B cells in lung tissue, prevents the formation of CS-induced LFs and the increase in immunoglobulin levels, and alleviates lung inflammation and alveolar wall destruction. 144
Additionally, inducible bronchus-associated lymphoid tissue (iBALT) plays a critical role in the pathogenesis of AECOPD. 144 iBALT contributes to the worsening of infection-related AECOPD. To further explore the role of iBALT in noninfectious AECOPD, researchers have found that oxidized sterols significantly modulate the formation of iBALT and the immunopathogenesis of COPD during chronic CS exposure. Elevated expression of CH25H and CYP7B1 in airway epithelial cells can regulate CS-induced B cell migration and iBALT formation. Treatment with clotrimazole, which inhibits the oxysterol pathway, effectively reduces iBALT formation and mitigates CS-induced emphysema. 146 Therefore, activation and formation of iBALT triggered by infection or certain noninfectious factors can exacerbate COPD, suggesting that targeting this pathway may offer new therapeutic strategies for AECOPD.
Antibodies are large Y-shaped proteins secreted by plasma cells (effector B cells) that the immune system uses to identify and neutralize foreign substances, such as bacteria and viruses. Based on the characteristics of B cells, monoclonal antibodies have been developed for the treatment of various diseases. These highly uniform antibodies are produced from a single B cell clone that targets specific antigen epitopes. COPD is typically characterized by type 2 (T2) inflammation. A clinical study observed significant improvements in lung function, sinus imaging, and serum T2 inflammatory markers in T2 inflammatory COPD patients treated with anti-IgE monoclonal antibodies. 147
Currently, molecular biology defines two opposing subpopulations of B cells based on the patterns of cytokine production. One is effector B cells (Befs), which actively regulate immune responses by releasing pro-inflammatory cytokines such as IL-6, IFN-γ, and GM-CSF. The other is regulatory B cells (Bregs), which negatively regulate immune responses through the release of anti-inflammatory cytokines like IL-10, IL-35, and transforming growth factor (TGF)-β. Bregs play a crucial role in maintaining immune balance by preventing excessive inflammation and tissue damage. A reduction or dysfunction of Bregs has been observed in various autoimmune diseases, infections, and cancers, leading to immune dysregulation. 148
Notably, compared to nonsmokers, both smokers without airflow limitation and patients with COPD exhibit a significantly lower percentage of IL-10+ Bregs in the blood-derived memory B cell subset. This suggests a potential link between reduced Breg numbers and function and the development and progression of AECOPD. 149 Further research into the specific mechanisms of action of Befs and Bregs may provide critical breakthroughs for the treatment of AECOPD.
Conclusion
AECOPD has caused great health and economic burden to society. The abuse of antibiotics and low efficiency are the main reasons. The root cause is the inaccuracy of treatment methods. Only by adopting different treatment methods for different causes in each immunization stage can the curative effect and quality of life of patients be effectively improved. Although many specific mechanisms have been found in the current research, the matching clinical diagnosis and treatment methods are still less applicable or lacking. Increasing the accurate diagnosis and treatment of patients with AECOPD as soon as possible is the top priority of clinical work at present.