
Abstract
Chronic obstructive pulmonary disease (COPD) is a complex chronic respiratory disease with cumulative impacts on multiple systems, exhibiting significant extrapulmonary impacts, and posing a serious public health problem. Skeletal muscle dysfunction is one of the most pronounced extrapulmonary effects in patients with COPD, which severely affects patient prognosis and mortality primarily through reduced productivity resulting from muscle structural and functional alterations. Although the detailed pathogenesis of COPD has not been fully determined, some researchers agree that oxidative stress plays a significant role. Oxidative stress not only catalyzes the progression of pulmonary symptoms but also drives the development of skeletal muscle dysfunction. Nuclear factor erythroid 2-related factor 2 (Nrf2), is a key transcription factor that regulates the antioxidant response and plays an enormous role in combating oxidative stress. In this review, we have summarized current research on oxidative stress damage to COPD skeletal muscle and analyzed the role of Nrf2 in improving skeletal muscle dysfunction in COPD through exercise. The results suggest that oxidative stress drives the occurrence and development of skeletal muscle dysfunction in COPD. Exercise may improve skeletal muscle dysfunction in patients with COPD by promoting the dissociation of Kelch-like ECH-associated protein 1 (Keap1) and Nrf2, inducing sequestosome1(p62) phosphorylation to bind with Keap1 competitively leading to Nrf2 stabilization and improving dynamin-related protein 1-dependent mitochondrial fission. Nrf2 may be a key target for exercise anti-oxidative stress to alleviate skeletal muscle dysfunction in COPD.
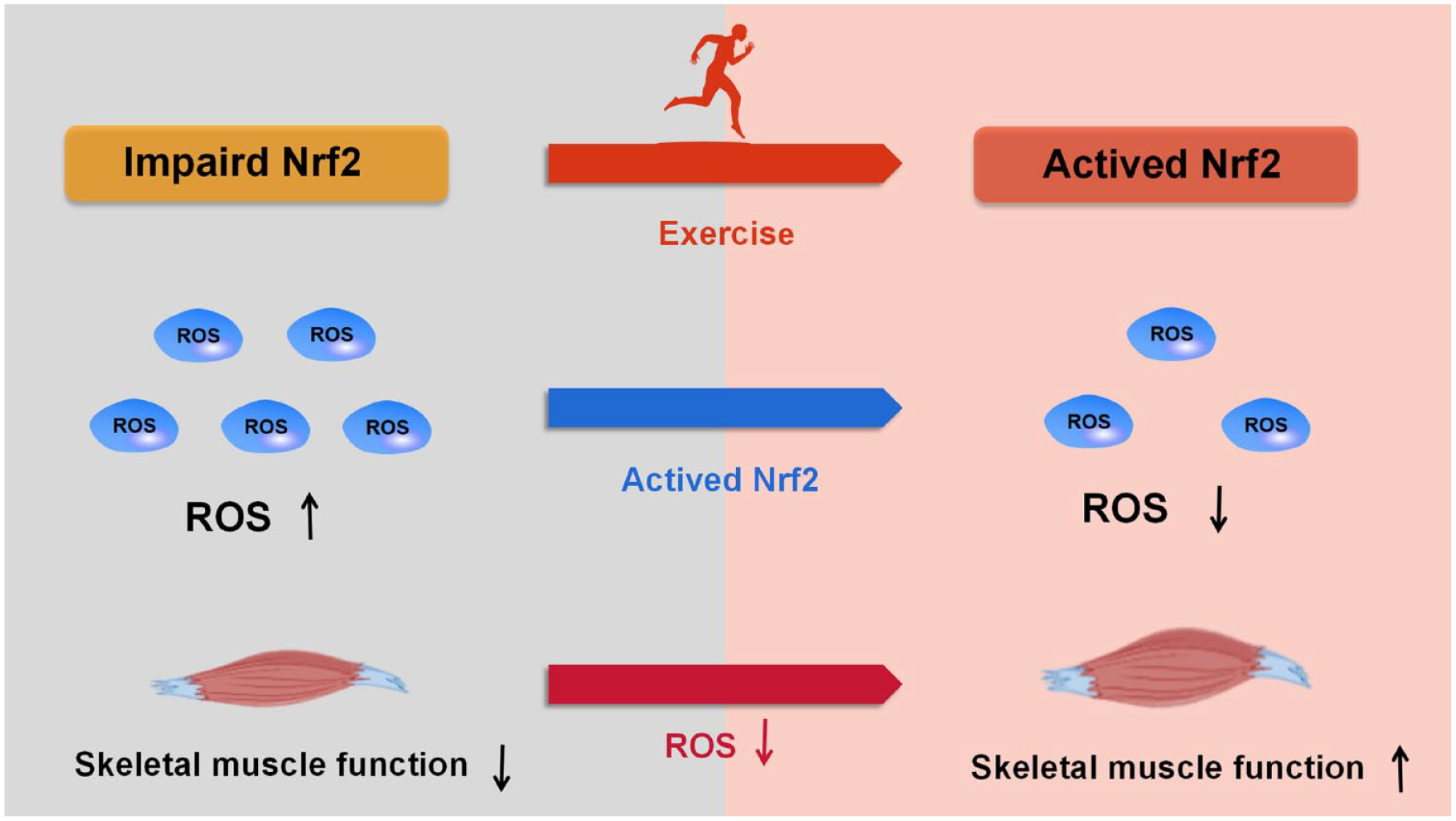
Introduction
Chronic obstructive pulmonary disease (COPD) is a significant and escalating global health concern, currently ranking as the third leading cause of death worldwide. 1 According to the Lancet, the number of people suffering from chronic respiratory disease was evaluated to be 544.9 million in 2017, in which approximately 55% of cases involve COPD. 2 COPD is a chronic lung disease characterized by airway and alveolar destruction that leads to persistent respiratory symptoms and not fully reversible airflow limitation. 1 Except for pulmonary alteration, COPD can also contribute to significant extrapulmonary effects. A primary and extremely prevalent extrapulmonary manifestation is skeletal muscle dysfunction, manifested as a decrease in muscle work efficiency and endurance due to structural and functional modification. 3 The structure is expressed as muscle atrophy, muscle fiber type shift, and reduction of muscle capillaries, and the function is characterized by a reduction of muscle strength and endurance. 4 The prevalence of skeletal muscle dysfunction in patients with COPD accounts for 32–33%, 5 which may appear in the mild stages of the disease, independently of lung function parameters, leading to decreased respiratory function and motor ability. 6 This parameter is an independent predictor of quality of life, cachexia, health care utilization, and mortality.7,8 Mitigating skeletal muscle dysfunction due to COPD represents a potentially novel orientation to improve the quality of life in patients with COPD, in contrast with addressing the primary pulmonary impairment of structure and function with COPD, which is not fully reversible. 6
Oxidative stress is an imbalance between oxidants and antioxidants in favor of the oxidants, leading to a disruption of redox signaling and control and/or molecular damage, 9 playing a crucial role in the progress of COPD skeletal muscle dysfunction. Oxidative stress in patients with COPD is concentrated not only in the lungs but also in skeletal muscles, and it is associated with a decrease in muscle strength. 10 Skeletal muscle dysfunction is more pronounced in exacerbating patients, 11 suggesting that oxidative stress is aggravated with disease progression. Nuclear factor erythroid 2-related factor 2 (Nrf2), a central transcription factor of the endogenous antioxidant pathway, plays a vital role in halting oxidation and maintaining the homeostasis of redox status in tissues and organs in response to oxidative and xenobiotic stress.12,13 Nrf2 inhibits age-related muscular dysfunction by activating antioxidant genes to release antioxidants against excessive reactive oxygen species (ROS) and restoring redox homeostasis. 14 Decreased Nrf2 expression in peripheral blood mononuclear cells derived from patients with COPD may be the consequence of a persistent oxidative attack on cellular defense mechanisms that exceeds their defense threshold, indicating the presence of Nrf2 antioxidant defense pathway disruption in COPD. 15 The activation of the Nrf2 signaling pathway against oxidative stress may be a potential biological target for the amelioration of COPD skeletal muscle dysfunction.
Exercise is an essential component of the management of COPD, and regular exercise can produce increases in the cross-sectional area, strength, and endurance of skeletal muscles, thereby promoting the daily activities and quality of life of patients.16,17 A profound comprehension of the mechanism through which exercise improves skeletal muscle dysfunction in patients with COPD could provide a theoretical foundation and fresh perspectives for preferably clinical management of patients with COPD. Exercise increases the antioxidant enzyme expression in the skeletal muscle of patients with COPD and lowers oxidative stress to improve 6-min walking test distance. 18 Multiple antioxidant enzyme expression requires the Nrf2 into the nucleus and binds to antioxidant response elements (ARE) to activate antioxidant genes. 19 The study has demonstrated that 9 weeks of resistance exercise can enhance Nrf2 and antioxidant enzyme expression in the skeletal muscle of rats with COPD skeletal muscle dysfunction to elevate endogenous antioxidant defense capacity. 20 In addition, supplementation with the Nrf2 activator sulforaphane enhances the benefits of exercise and exerts a protective effect on muscle by reducing oxidative stress to skeletal muscle in mice. 21 Hence, Nrf2 performs a vital role in exercise to increase muscle antioxidant capability to ameliorate skeletal muscle dysfunction. This article aims to review the role of oxidative stress in COPD skeletal muscle dysfunction and to investigate the contribution of Nrf2 in the amelioration of oxidative stress in COPD skeletal muscle by exercise.
Skeletal muscle dysfunction in COPD
Skeletal muscle dysfunction, a frequent secondary systemic manifestation in patients with COPD, is characterized by decreased muscle strength and/or endurance 22 that affects respiratory function and is also associated with poor exercise performance, fatigue, and increased risk of fracture. 23
Structural modifications of skeletal muscle in COPD
Structural alterations of skeletal muscle in COPD include atrophy, fiber type shift, and reduction of muscle capillaries. Skeletal muscle atrophy is primarily characterized by a reduction in cross-sectional area and muscle density involving all muscle fiber types, 24 which can be more serious than the atrophy caused by normal aging. 25 This condition frequently occurs in patients with low body weight and can also be present in patients with normal body mass index. 26 The prevalence rate of muscle atrophy in COPD is in the range of 4–35%, identified as in the disease’s early stage, 27 and its onset correlates to severity.28,29 At an equal level of airway obstruction, emphysematous phenotype patients have relatively more muscle wasting than patients with chronic bronchitis and muscle wasting is accelerated during acute disease exacerbations, but few studies have reported muscle loss trajectories. 30 Different muscle groups present variables in COPD; for example, type I fiber proportions are markedly low, and type II fiber proportions remarkably high in the vastus lateralis muscle, but opposite changes to these above-mentioned have been observed in the inspiratory muscle.31,32 Type I fibers have better oxidative metabolism, calcium sensitivity, and fatigue resistance than type II fibers.33–35 The transformation of muscle fiber type resulted in a decline in the oxidative capacity and fatigue resistance of the peripheral muscles and an elevation of these in diaphragm. Peripheral muscle capillary density and capillary-to-fiber ratio decreased in patients with COPD than in controls,24,36 but diaphragm capillary density and the number of vessels per muscle fiber increased significantly. 37 Capillaries play a major role in the metabolic homeostasis of skeletal muscles and are essential pathways for the exchange of oxygen, substrates, and metabolites between blood and cells. 38 The diffusion of oxygen to skeletal muscle is primarily dependent on the quantity of capillaries and the number of blood vessels per muscle fiber, which directly determines the rate of oxygen delivery from the blood to the muscle and thus the efficiency of muscle work. 39 The modifications in capillary density and the number of vessels per muscle fiber in the peripheral muscles and diaphragm of patients with COPD result in a decrease in the capacity for oxygen exchange in the peripheral muscles and that increase in diaphragm, which is similar to the consequences of the fiber type shift. This heterogeneous switch in muscle fiber type and variation in capillary density in the diaphragm from the peripheral muscle responds to the endurance training-like effect caused by increased respiratory work in patients with COPD. 40
Function modification of skeletal muscle in COPD
The changes in skeletal muscle function in COPD are mainly reflected in the reduction of muscle strength and endurance. The skeletal muscle strength decreases during the late stages of the disease but can also emerge in the early stages of the disease, and the prevalence of quadriceps weakness has reached approximately 20% even in patients with mild COPD. 5 A considerable variability has been observed among patients, with some patients having relatively normal values, whereas others have a reduction in strength of more than 50%. 41 The deterioration of muscle strength in patients with COPD involves the diaphragm, quadriceps, deltoid, and latissimus dorsi, and the reduction is more significant in the lower than in upper extremities, 42 suggesting that local factors are critical in regulating the phenotype of skeletal muscle-related changes in COPD. Volitional 43 and non-volitional 44 muscular endurance assessments both indicated that skeletal muscle endurance was significantly reduced, and fatigue was more frequent in patients with COPD than age-matched healthy individuals.45,46 The prevalence of decreased skeletal muscle endurance in COPD ranges widely from approximately 32–77% considering the differences in test procedures including the type of muscle contraction, velocity of exercise and equipment used at the moment of measurement. 28 Declines in skeletal muscle endurance have been observed in patients with severe COPD, as well as those suffering from mild to moderate COPD,47,48 and the extent of the endurance decline may be greater than the muscle strength, 49 implicating that muscle endurance training should not be neglected in the COPD training program.
Oxidative stress and COPD skeletal muscle dysfunction
Oxidative stress is a critical pathogenic mechanism of COPD skeletal muscle dysfunction
Oxidative stress is defined as a disequilibrium between production of free radicals and ROS in favor of oxidation, and their elimination, considering the impaired or overloaded protective mechanisms. 50 ROS are oxygen centered radicals or non-radical reactive derivatives of oxygen molecules, which react with a wide spectrum of molecules to modify their structure or function in reversible or irreversible manners. 51 Although excessive ROS is harmful by nature, they are fundamentally important for many physiological processes as functional signaling entities. 52 Under physiological status, ROS are constantly produced at a low level in living systems, but the generation is antagonized by cellular antioxidant activity, which functions to prevent, delay, or remove oxidative damage caused by biological molecules. 53 Intracellular redox homeostasis is crucial for maintaining healthy skeletal muscle. However, when oxidative stress occurs due to redox homeostasis in favor of the oxidants, important biomolecules such as proteins, lipids, and DNA are subject to oxidative damage and thus potentially adversely affect the organism as a whole. 54
Oxidative stress is the main actuating mechanism of COPD, and it is tightly connected with nuclear respiratory symptoms (cough, expectoration, and difficulty breathing) and influences skeletal muscle. 55 In COPD, local56,57 and systemic 58 oxidative stress have been reported. Oxidative stress has long been implicated in the genesis of sarcopenia, particularly in patients with severe symptoms. 59 Oxidative stress can induce cell destruction and death, impair myogenic differentiation, 60 activate the ubiquitin protease system, and disrupt protein synthesis, 61 all of which may predispose to muscle atrophy. Several investigations have consistently demonstrated that under resting and exercise conditions, patients with COPD exhibit increased levels of lipid peroxidation, oxidized glutathione, and protein carbonylation and nitration in their blood and both respiratory and peripheral muscles. 62 Systemic and local oxidative stress in patients with COPD correlated negatively with fat free mass and muscle strength, suggesting a link between oxidative stress and loss of muscle mass. 63 In addition, oxidative stress levels contribute to the regulation of autophagy, which is involved in the atrophy of COPD skeletal muscle. 64 Therefore, oxidative stress plays a significant role in the initiation and prognosis of skeletal muscle dysfunction in COPD.
Nrf2 and COPD skeletal muscle oxidative stress
Nrf2 mediates the emergence of phase II antioxidant genes encoding cellular defense and protection in response to oxidative stress. 65 As a predominant transcription factor, Nrf2 moderates more than 200 cytoprotective genes to cope with oxidative stress such as homeostasis 1 (HO-1), glutathione, quinone oxidoreductase, and superoxide dismutase (SOD), and it plays a central role in optimizing intracellular redox.66–68 Under quiescent/homeostatic/baseline conditions, most Nrf2 continuous combinates with the Kelch-like ECH-associated protein 1 (Keap1), an adaptor subunit for Cullin3-based ubiquitin E3 ligase,69,70 leading to a low concentration of Nrf2 in the cytoplasm. This E3 ligase complex efficiently ubiquitinates Nrf2, leading to its rapid proteasomal degradation. 71 However, under oxidative stress conditions, increased ROS induces the dissociation of Keap1 from Nrf2 in response to oxidative stress, and Nrf2 is stabilized by dissociating from Keap1, permitting the translocation of Nrf2 into the nucleus, binding to cis-elements called ARE as a heterodimer with other members of the basic leucine zipper protein family, and activating a large group of antioxidant-associated genes to restore redox balance. 72
Various studies have confirmed the association of Nrf2 with numerous lung diseases, including COPD. Macrophages in lung of elderly smokers and patients with COPD exhibit downregulated expression of Nrf2 mRNA. 73 The expression of Nrf2 and Nrf2-related genes HO-1 and glutamate-cysteine ligase catalytic subunits is diminished in peripheral blood mononuclear cells of patients with COPD. 15 The Nrf2 signaling pathway partially preserves the structure and function of skeletal muscle in mice by maintaining redox homeostasis. 74 Nrf2-deficient mice exhibit increased skeletal muscle ROS production under basal conditions, thereby disrupting antioxidant defense responses and rendering the skeletal muscle dysfunctional owing to oxidative stress. 75 This process may enhance oxidative modification of structural and functional proteins in skeletal muscle cells. 76 By contrast, the up-regulation of Nrf2 protein has boosted exercise endurance and alleviated fatigue in overworked mice, 77 illustrating that Nrf2 is essential for maintaining healthy skeletal muscle function. No significant differences were observed in the expression of the Nrf2 mRNA of quadriceps muscle of mice with skeletal muscle dysfunction induced by 12-week cigarette smoke exposure, but the expression of Keap1 mRNA was clearly elevated. 78 In the diaphragm of smoke-exposed mice at 12 weeks, Nrf2 mRNA presentation decreased, and HO-1 mRNA remarkably decreased compared with the controls. 79 Nrf2, HO-1 and quinoneoxido-reductase-1 (NQO-1) transcript levels were decreased in C2C12 cells by using serum starvation-induced myotubular atrophy, and oxidative stress-induced myotubular atrophy was mitigated by the upregulation of Nrf2-mediated antioxidation. 80 The above studies indicate that Nrf2 may occupy an important position in counteracting oxidative stress to preserve skeletal function in COPD.
Effects of exercise on skeletal muscle function and Nrf2 pathway in COPD
Exercise, a full-fledged component of pulmonary rehabilitation, is the most significant intervention currently available for the treatment of skeletal muscle dysfunction in patients with COPD. 81 Exercise boosts the cross-sectional area of skeletal muscle to improve exercise performance and endurance for COPD, and it is a remarkable non-pharmacological treatment strategy for controlling muscle oxidative stress and inflammation. 82 Evidence from animal and human studies indicate that the benefits of exercise can be attributed to redox signaling cascades that are mediated by Nrf2 signaling.83,84 Exercise can activate Nrf2 and downstream target genes to ameliorate COPD skeletal muscle dysfunction, and the detailed literature is shown in Table 1. Exercise training facilitates the dissociation of Keap1 and Nrf2 in COPD skeletal muscle, p62 phosphorylation induces Nrf2 nuclear translocation to activate antioxidant genes and increases dynamin-related protein 1 (Drp1) stabilization in order to enhance Drp1-dependent mitochondrial fission to improve COPD skeletal muscle dysfunction as detailed in Figure 1.
Effects of exercise on Nrf2 and targeted genes of skeletal muscle in COPD.

↑: The value of exercise + COPD group/exercise+cigarette smoking was significantly higher than that of COPD group/cigarette smoking group or baseline. ↓: The value of exercise +COPD was significantly lower than that of COPD group or baseline.
6MWT, 6-min walking test; COPD, chronic obstructive pulmonary disease; ET, endurance training; G6PD, glucose-6-phosphate dehydrogenase; GSH, Glutathione; GSR, glutathione reductase; HIIT, high-intensity interval training; HO-1, heme oxygenase 1; NAD(P)H, quinone oxidoreductase 1; Keap1, Kelch-like ECH-associated protein 1; NOX, NADPH oxidase; NQO-1, quinoneoxido-reductase-1; Nrf2, nuclear factor, erythroid derived 2, like 2; Trx, thioredoxin; TrxR: thioredoxin reductase; SOD, superoxide dismutase; RM, repetition maximum; RT, resistance training; RWpeak, peak work rate; VO2peak, peak oxygen uptake.

Exercise activates Nrf2 to improve skeletal muscle dysfunction in COPD.
Exercise suppresses Keap1 to activate Nrf2
An important mechanism regulating Nrf2 activation is the inhibition of Keap1 activity, given that the conventional mechanism controlling Nrf2 stability is mediated by Keap1. Toledo-Arruda et al. 78 discovered that 12 weeks of aerobic training counteracted weight loss, the decline in exercise capacity, the elevation of thiobarbituric acid reactive substances and protein carbonylation as oxidative stress markers caused by cigarette smoke exposure in mice. Meanwhile, quantitative real-time PCR (RT-PCR) revealed that this training elevated the mRNA abundance of Nrf2 and its target genes glutathione reductase and thioredoxin, while decreasing the mRNA expression of Keap1 in the quadriceps muscle. 78 Nonetheless, 4 and 8 weeks of aerobic exercise did not provoke any alteration of Nrf2 mRNA in mice quadriceps muscle, 78 revealing that the activation of Nrf2 by exercise training is dependent on the duration of exercise. Yahui 20 found that 9 weeks of resistance exercise enhanced the cross-sectional area and motility of skeletal muscle in rats with COPD skeletal muscle dysfunction. Additionally, this exercise simultaneously increased the protein content of Nrf2 and the antioxidant protein SOD1 and diminished the amount of Keap1 protein in skeletal muscle measured by Western blot. 20 Based on the present results, exercise may lower Nrf2 degradation by suppressing Keap1 performance and activate endogenous antioxidant defense by accumulating Nrf2 in the cytoplasm and then in the nucleus, thus ameliorating skeletal muscle function. Aerobic exercise for 24 weeks elevated Nrf2 mRNA presentation in the diaphragm of mice with COPD diaphragm dysfunction, and unlike the results of the above study, the expression of the Keap1 gene increased. 79 Keap1 gene expression variation induced by exercise duration may be explained by the existence of a counter-regulatory mechanism between Keap1 and Nrf2.
Exercise promotes p62 phosphorylation to induce Nrf2 nuclear translocation
Sequestosome1(p62/SQSTM1) is an autophagic connector protein engaged in various cellular functions, ranging from signals transduction to the degradation of proteins and organelles.87,88 The phosphorylation of p62 increases its affinity for Keap1, competes for binding between Keap1 and Nrf2 and thus leads to dissociation of Nrf2 and Keap1, increasing Nrf2 activation and nuclear translocation,89,90 suggesting that p62 exerts a regulatory role in the performance of Nrf2. Further investigation revealed that p62 is engaged in the regulation of Nrf2 during exercise, given that exercise elevates the phosphorylation level of p62 in the lateral femoral muscle of actively exercising men, accompanied by an increase in phosphorylated Nrf2 and a decrease in the abundance of Keap1 protein. 91 Yamada et al. 92 found that 4 weeks of autonomous running wheel training enhanced Nrf2, p62, phosphorylated p62, antioxidant enzyme SOD1, SOD2, SOD3, and NQO-1 protein abundance in mice soleus muscles measured by Western blot, and p62 knockout mice exhibited the same impaired exercise-mediated increase in SOD1 and SOD3 expression in skeletal muscle as Nrf2 knockout mice. Therefore, the exercise-mediated growth of antioxidant enzymes may be mediated by p62 phosphorylation competing for Keap1 and Nrf2 binding to contribute to Nrf2 accumulation and nucleation. Exercise may promote p62 phosphorylation that competes with Keap1 and Nrf2 binding to induce Nrf2 accumulation and nucleation, activating downstream antioxidant genes that target oxidative stress to alleviate skeletal muscle dysfunction in patients with COPD.
Exercise activates Nrf2 to increase Drp1 stabilization and thus enhance Drp1-dependent mitochondrial fission
Mitochondria are the predominant origin of intracellular ROS, 93 and the ROS produced in mitochondria can trigger mitochondrial dysfunction through interactions with mitochondrial and cellular components, including reduced mitochondrial biogenesis, altered membrane potential, diminished mitochondrial number, and altered activity of oxidative proteins. 94 The mitochondrial dysfunction can cause a disequilibrium between ROS production and elimination, which can result in an elevated ROS.95,96 Mitochondrial dysfunction is an overwhelming potential mechanism of skeletal muscle dysfunction. In comparison to wild-type aged mice, Nrf2 knockout aged mice exhibited reduced antioxidant genes expression and elevated mitochondrial markers of oxidative damage, 97 indicating that Nrf2 plays a crucial role in regulating oxidative homeostasis and maintaining healthy mitochondrial function. Drp1 is the major pro-fission protein with strictly controlled activity and acts as a major role in maintaining mitochondrial fusion and fission. 98 The evidence for Drp1 regulating exercise performance and exercise adaptation is clear, mice with specific deletion of skeletal muscle Drp1 exhibited decreased exercise tolerance, maximal running speed, and altered muscle adaptations in response to endurance exercise. 99 Therefore, Drp1 has an instrumental effect on the retention of normalized skeletal muscle function and the associated improvement in endurance from exercise training. RT-PCR results showed the mRNA expression levels of Nrf2 and Drp1 increased in skeletal muscle after acute medium-intensity endurance and high-intensity intermittent exercise. 100 Yan et al. 101 found that an 8-week exercise intervention elevated Nrf2 mRNA in the skeletal muscle of aged mice revealed by RT-PCR, and Nrf2 increased Drp1 stability through deubiquitination and promoted Drp1-dependent mitochondrial fission. The generalization of mitochondrial fission dependent on Drp1 helps reduce abnormal mitochondrial swelling and reduced mitochondrial biogenesis 102 to maintain the health of aging skeletal muscle. An alleviated mitochondrial function may correct the inequality between ROS production and elimination and thus ameliorate oxidative stress. COPD, one of the diseases closely associated with aging, increases significantly in prevalence with age, 103 with a five-fold higher risk of developing COPD at age 65 compared to patients under 40 years. 104 Based on the finding above, exercise can activate Nrf2 to increase Drp1 stabilization and reduce mitochondrial dysfunction to strengthen skeletal muscle motility in COPD, but the exact mechanism remains to be confirmed.
Conclusion
Skeletal muscle dysfunction has become a prevalent and grievous extrapulmonary phenomenon in patients with COPD and can be an independent predictor of prognosis in patients with COPD independently of their respiratory symptoms. Regional and systemic oxidative stress are pivotal trigger mechanisms for the development of skeletal muscle dysfunction in patients with COPD, and the intersection between Nrf2 and oxidative stress is needed to maintain skeletal muscle function by combating oxidative stress. As a dominant intervention for skeletal muscle dysfunction in COPD, both aerobic and resistance exercise can activate the Nrf2 signaling pathway in the skeletal muscle of patients with COPD, and the activation of the Nrf2 signaling pathway is dependent on the duration of exercise. Exercise may activate Nrf2 by inhibiting Keap1 to activate Nrf2, promoting p62 phosphorylation to trigger Nrf2 nuclear translocation, and activating Nrf2 to decrease abnormal mitochondrial swelling and biogenesis by increasing Drp1 stabilization.
Limitations and perspectives
Anti-oxidative stress-based therapy is a new therapeutic target to ameliorate skeletal muscle dysfunction in COPD. Nonetheless, there is still limited understanding of various aspects of exercise in terms of activating Nrf2 to alleviate skeletal muscle dysfunction in patients with COPD. Currently, the effect of exercise on Nrf2 in COPD skeletal muscle is still in the animal research stage. Existing studies have focused on the effects of exercise on Nrf2 protein and mRNA expression levels in COPD skeletal muscle, but the effects of exercise on Nrf2 knockout/knockdown or overexpressed COPD skeletal muscle function and oxidative stress have not been explored. The following direction aims to investigate the contributions of different exercise durations, exercise modes, the exercise intensity to the Nrf2 signaling pathway in COPD skeletal muscle dysfunction, the effects of the same exercise prescription on the Nrf2 pathway in different muscle groups, and the influences of disease severity and phenotype on the activation of the Nrf2 signaling pathway by exercise to provide novel insights and rationales for the formulation of exercise rehabilitation prescriptions for clinical COPD skeletal muscle dysfunction. At the same time, they aim to further explore the molecular mechanisms of exercise to activate the skeletal muscle Nrf2 signaling pathway in COPD skeletal muscle dysfunction and establish the theoretical basis for exercise training targeting the Nrf2 signaling pathway to counteract oxidative stress and improve COPD skeletal muscle dysfunction.