
Abstract
Background and objectives:
eNAMPT (extracellular nicotinamide phosphoribosyltransferase), a novel DAMP and TLR4 ligand, is a druggable ARDS therapeutic target with NAMPT promoter SNPs associated with ARDS severity. This study assesses the previously unknown influence of NAMPT promoter SNPs on NAMPT transcription, eNAMPT secretion, and ARDS severity.
Methods and design:
Human lung endothelial cells (ECs) transfected with NAMPT promoter luciferase reporters harboring SNPs G-1535A, A-1001 C, and C-948A, were exposed to LPS or LPS/18% cyclic stretch (CS) and NAMPT promoter activity, NAMPT protein expression, and secretion assessed. NAMPT genotypes and eNAMPT plasma measurements (Days 0/7) were assessed in two ARDS cohorts (DISCOVERY n = 428; ALVEOLI n = 103).
Results:
Comparisons of minor allelic frequency (MAF) in both ARDS cohorts with the 1000 Human Genome Project revealed the G-1535A and C-948A SNPs to be significantly associated with ARDS in Blacks compared with controls and trended toward significance in non-Hispanic Whites. LPS-challenged and LPS/18% CS–challenged EC harboring the -1535G wild-type allele exhibited significantly increased NAMPT promoter activity (compared with -1535A) with the -1535G/-948A diplotype exhibiting significantly increased NAMPT promoter activity, NAMPT protein expression, and eNAMPT secretion compared with the -1535A/-948 C diplotype. Highly significant increases in Day 0 eNAMPT plasma values were observed in both DISCOVERY and ALVEOLI ARDS cohorts (compared with healthy controls). Among subjects surviving to Day 7, Day 7 eNAMPT values were significantly increased in Day 28 non-survivors versus survivors. The protective -1535A SNP allele drove -1535A/-1001A and -1535A/-948 C diplotypes that confer significantly reduced ARDS risk (compared with -1535G, -1535G/-1001 C, -1535G/-948A), particularly in Black ARDS subjects. NAMPT SNP comparisons within the two ARDS cohorts did not identify significant association with either APACHE III scores or plasma eNAMPT levels.
Conclusion:
NAMPT SNPs influence promoter activity, eNAMPT protein expression/secretion, plasma eNAMPT levels, and ARDS severity. NAMPT genotypes are a potential tool for stratification in eNAMPT-focused ARDS clinical trials.
Keywords
Introduction
Acute respiratory distress syndrome (ARDS) is triggered by diverse direct and indirect injurious challenges to the lung that result in diffuse inflammatory injury and loss of alveolar and vascular lung barrier integrity. 1 Prior to the COVID-19 pandemic, US ARDS cases numbered over 200,000 persons annually with 2 million cases globally. Mortality rates exceed 30–40%, 1 and since the onset of the COVID-19 pandemic, ARDS deaths worldwide have skyrocketed with ARDS being a primary cause of mortality in SAR-CoV2-infected individuals. 2 The absence of effective Food and Drug Administration (FDA)-approved ARDS therapeutics is a serious and sobering unmet need highlighted by the COVID-19 pandemic with numerous failed ARDS clinical trials, reflecting the heterogeneity of the disease and the absence of detailed phenotyping for ARDS subjects at trial enrollment. Criteria for ARDS severity, such as the Berlin criteria for ARDS severity (mild, moderate, severe ARDS),3,4 are significantly predictive of survival; however, a subphenotyping tool with therapeutic utility has yet to emerge. A clinically useful biomarker with the potential to identify specific ARDS subphenotypes and/or therapeutic targets would be significantly beneficial in promoting effective clinical trial designs to assess novel therapeutics. 5
We have previously utilized genomic-intensive approaches to identify potentially novel therapeutic targets in acute inflammatory lung disorders including ARDS and ventilator-induced lung injury,6–10 studies that identified NAMPT as a novel candidate gene in ARDS.10–12 NAMPT encodes extracellular nicotinamide phosphoribosyltransferase (eNAMPT), which, similar to the high motility group box 1 (HMGB1) and macrophage migration inhibitory factor (MIF) 7 , is a dual-functioning cytozyme with intracellular enzymatic activity involved in nicotinamide adenine dinucleotide (NAD) biosynthesis (iNAMPT) and extracellular inflammatory cytokine activity (eNAMPT). We were the first to demonstrate that secreted eNAMPT, like HMGB1, is a damage-associated molecular pattern protein (DAMP) and ligand for Toll-like receptor 4 (TLR4), 13 potently stimulating dysregulated inflammatory responses that contribute to lung inflammation. Nampt+/– heterozygous mice are protected from bacterial- and ventilator-induced lung injury (VILI), 14 and circulating eNAMPT directly contributes to preclinical lipopolysaccharide (LPS)/VILI-induced lung injury in mouse, rat, and porcine models as validated by studies utilizing a humanized eNAMPT-neutralizing mAb.14–16 The protein levels of eNAMPT are significantly increased in the bronchoalveolar lavage fluid and blood from ARDS subjects,10,17–20 including COVID-19-infected subjects. 21 The association of eNAMPT blood levels with ARDS severity provides support for plasma eNAMPT as a potentially clinically-relevant biomarker in ARDS.
Additional compelling support for eNAMPT as an important contributor to ARDS pathobiology is derived from the observation that NAMPT promoter single nucleotide polymorphisms (SNPs) are associated with ARDS risk, severity, and mortality.10,12,22,23 Our prior mechanistic studies to interrogate NAMPT promoter regulation identified specific transcription factors (STAT5, HIF-2α, SOX17, SOX18)12,24,25 that regulate NAMPT promoter activity in response to inflammatory stimuli, hypoxia, and mechanical stress (18% cyclic stretch or CS).10,12,26 The NAMPT gene also undergoes epigenetic regulation via microRNAs (miRNAs) and by promoter CpG demethylation induced by 18% CS or LPS. 24 NAMPT promoter SNPs at -1535 and -1001 (from TSS) have been linked to ARDS risk and mortality.10,22,23 The NAMPT-948 SNP was also reported to alter transcription factor binding.12,24,25
Given the participation of the highly targetable eNAMPT/TLR4 inflammatory signaling pathway in ARDS,15,16,27 we hypothesized that exploration of the genotype–phenotype link between NAMPT promoter SNPs, plasma eNAMPT biomarker levels, and ARDS severity may provide clinically useful insights that may be incorporated into ARDS clinical trial design. This study, using in vitro techniques and biochemical/genetic analyses of samples from well-phenotyped ARDS cohorts, demonstrates that specific NAMPT promoter SNPs regulate NAMPT promoter activity and potentially influence eNAMPT secretion into the circulation and ARDS severity. Although these studies were significantly underpowered, eNAMPT plasma levels and the risk NAMPT genotype appear to represent novel stratification tools to increase the success of eNAMPT-focused therapeutic clinical trials in high-risk ARDS subjects.
Methods
Demographics of ARDS genotyping and biomarker cohorts
A total of 474 ARDS subjects comprised the ARDS DISCOVERY cohort (University of Arizona IRB #CR00000082 and University of Illinois #20120192), representing six distinct cohorts including a National Institutes of Health (NIH) cohort (BioLINCC request #9578) ‘Fluid and Catheter Treatment Trial’ (FACTT, 203 subjects). 20 The characteristics of these cohorts are presented in Supplemental Table 1. Selected specimens based on availability were obtained from a second NIH ARDS cohort (BioLINCC request #12130), ‘Assessment of Low tidal Volume and Elevated End-Expiratory Volume to Obviate Lung Injury’ (ALVEOLI) trial (103 subjects from a 525 subject cohort). Both FACTT and ALVEOLI are multi-centered randomized controlled trials performed by the National Heart, Lung, and Blood Institute (NHLBI) ARDSNet conducted at multiple academic health centers and previously described.28,29 The ARDS diagnosis was established following the Berlin Criteria. 4 The 28-day mortality outcomes obtained on each ARDS cohort are reported in Tables 1 and 2. The APACHE (Acute Physiology And Chronic Health Evaluation) score, a series of severity-of-disease classifications used in the intensive care unit (ICU), 30 was utilized to quantify risk (higher score equates to higher risk of mortality). In the discovery cohort, both APACHE II scores (n = 150) and APACHE III scores were utilized (n = 314) (Table 1). The ALVEOLI cohort (n = 103) reported APACHE III scores (Table 2). Two independent cohorts of healthy donors [University of Arizona institutional review board (IRB) #1312168664R001] were assessed for plasma eNAMPT levels using either the in-house enzyme-linked immunosorbent assay (ELISA, n = 268) or the electrochemiluminescence multiplex immunoassay (n = 78). All data were completely deidentified to avoid potential sources of bias.
Characteristics of the ARDS DISCOVERY cohort by survival.

APACHE, Acute Physiology and Chronic Health Evaluation; ARDS, acute respiratory distress syndrome; eNAMPT, extracellular nicotinamide phosphoribosyltransferase.
Pearson’s chi-square test.
Two-sample t test with equal variance.
Pearson’s chi-square test comparing African and European self-report descent between outcome conditions.
Utilizing the in-house ELISA assay.
Self-reported race.
Characteristics of the ALVEOLI ARDS cohort (n = 103).

ALVEOLI, Assessment of Low tidal Volume and Elevated End-Expiratory Volume to Obviate Lung Injury; APACHE, Acute Physiology and Chronic Health Evaluation; ARDS, acute respiratory distress syndrome; eNAMPT, extracellular nicotinamide phosphoribosyltransferase; MSD, Meso Scale Discovery.
Utilizing the MSD ELISA platform.
DNA subject sample collection
For the ARDS DISCOVERY cohort, DNA was derived from peripheral blood mononuclear cells (PBMCs) and collected in accordance with their respective IRBs and transferred with appropriate material transfer agreements. For ALVEOLI samples, cell-free DNA was extracted from plasma (200 μl) using the Quick-cfDNA Serum & Plasma Kit (Zymo Research, Irvine, CA, USA) following the manufacturer’s instructions. The extracted DNA was stored in RNA later or Triazol according to the standard protocol.
eNAMPT measurements
Blood from subjects in both ARDS cohorts was collected in ethylenediaminetetraacetic acid (EDTA)-treated tubes within 36 h of ARDS onset defined as when all Berlin ARDS Criteria were met 4 and platelet-depleted plasma stored at −80°C. Subjects were included if two time points were available with the first specimen was defined as D0 collection and the second specimen collected 7 days later (D7). For the ARDS DISCOVERY cohort, a 50-µl aliquot of plasma was utilized for an in-house ELISA to quantify plasma levels of eNAMPT as previously described18,31 utilizing a proprietary polyclonal goat anti-NAMPT antibody, human rhNAMPT, a polyclonal rabbit anti-NAMPT antibody (Bethyl Laboratories, Montgomery, TX, USA), and a secondary donkey anti-rabbit horseradish peroxidase (HRP)-labeled antibody (Life Technologies/ThermoFisher). The plates were read at 490 nm on Bio-Rad iMARK (Hercules, CA, USA) and the standardization of results previously described.18,25,31 For measurements of eNAMPT levels in plasma obtained from ALVEOLI subjects or in human endothelial cell (EC) supernatants, 50 µl of either plasma or EC supernatant was utilized in an electrochemiluminescence multiplex immunoassay predesigned panel (Meso Scale Discovery, MSD®) as we previously reported.21,27
NAMPT genotyping
Three NAMPT SNPs targeted for genotyping were selected on the basis of CpG sites, transcription factor–binding sites, minor allele frequency in populations with European and African ancestry, and known associations with ARDS risk.10,12,22–24 The three NAMPT SNPs are shown in Figure 1(a) and were genotyped either using the Agilent MassArray platform (Agena Bioscience, San Diego, CA, USA) (n = 474) or the N-Plex platform (MSD) (n = 103). Each SNP passed genotyping quality control (QC metrics included genotyping success rate of >90% per SNP, >50% per individual, and Hardy–Weinberg equilibrium (p > 0.05). Of the 474 DISCOVERY cohort ARDS subjects, 428 subjects had available outcome data for 28-day mortality. Twenty-three DNA samples failed genotyping of at least one SNP, yielding a total number of 405 ARDS genotyped subjects with mortality data. NAMPT genotyping of the 103 ARDS ALVEOLI cohort subjects utilized the N-Plex platform (MSD). Briefly, specimens were multiplexed and polymerase chain reaction (PCR)-amplified, followed by an Oligo Ligation Assay (OLA), Taq DNA ligase reaction for upstream and downstream probes, followed by OLA product hybridization and SULFO-TAG ™ detection, and plate reads with the MSD instrument.

Effect of NAMPT SNPs on human endothelial cell promoter activity and functional responses. (a) Shown is the schematic of the NAMPT promoter (-3028 to transcription start site at +1) with the three promoter SNPs depicted. (b) and (c) NAMPT promoter reporter plasmids were constructed by the NAMPT promoter region (−3028 bp to +1 ATG) and pGL3-basic vector (Promega Co.). The SNPs were generated by site-directed mutagenesis (QuikChange Lightning Multi Site-Directed Mutagenesis Kit; Agilent Inc, Santa Clara, CA, USA). ECs transfected with the NAMPT luciferase promoter harboring the -1535A NAMPT SNP demonstrated attenuated increases in NAMPT promoter activity in response to exposure to LPS (for 4 h, p < 0.008) (b), 18% cyclic stretch (CS, 4 h) (c), or combined and LPS/18% CS exposure (8 h, p < 0.001) (c) compared with -1535G wild-type allele controls. (d) Luciferase reporter activity is reduced in EC transfected with NAMPT luciferase promoter harboring the -1535A/-946 C diplotype compared with the -1535G/-948A diplotype, either at baseline or after LPS, 18% CS, or LPS/18% CS. (e) and (f) NAMPT protein expression in EC transfected with NAMPT plasmids with either the -1535A/-948 C or -1535G/-948A diplotypes in response to exposure to LPS was measured by immunoblotting EC lysates from EC transfected with -1535A/-948 C. These studies show attenuated increases in NAMPT protein expression in response to exposure to LPS in EC transfected with 1535A/-948 C versus -1535G/-948A (p < 0.05). NAMPT expression was confirmed by densitometric quantification by ImageJ. (g) eNAMPT secretion from lung EC was measured and cell supernatants from transfected EC (1535A/-948 C versus -1535G/-948A) by MSD ELISA. eNAMPT secretion was significantly increased in EC-overexpressing NAMPT under control of either NAMPT promoter. However, EC with the -1535G/-948A diplotype demonstrated significantly increased eNAMPT secretion compared with EC transfected with the -1535A/-948 C diplotype. (h) Effects of NAMPT overexpression on EC barrier function measured by transendothelial electrical resistance (TER). Overexpression of NAMPT with both -1535A/-948 C and -1535G/-948A significantly augmented thrombin-induced EC permeability. However, -1535A/-946 C diplotype showed significantly less EC barrier integrity loss in response to thrombin, compared with EC transfected with the gene under control of the NAMPT promoter with the -1535G/-948A diplotype (p < 0.05).
Human lung EC culture
Human lung ECs (Lonza Inc., Allendale, NJ, USA) were cultured as previously described 31 and grown to 70–75% confluence for transfection with either pGL3 sham vector or -1535A vector. Sham and -1535A-transfected EC were challenged with LPS (100 ng/ml, 18 h) with or without an additional 4-h exposure to 18% CS (0.5 Hz, FX-5000 System; FlexCell International, Hillsborough, NC, USA) to mimic high-tidal-volume mechanical ventilation as we previously described.12,24–26
Gene cloning and mutagenesis
Gene cloning and mutagenesis were performed as previously described. 12 Briefly, 3028 bp DNA fragments of the NAMPT promoter region (−3028 bp to +1 ATG) were amplified by PCR using the human genomic DNA as template and modified by site-directed mutagenesis to generate fragments containing minor alleles of NAMPT SNPs at these loci. All allele-containing fragments were fused to a pGL3-basic reporter vector (Promega, Madison, WI, USA). To further study the effects of genetic variants on gene expression, the NAMPT promoter harboring specific SNPs was subcloned and inserted upstream of NAMPT gene in pCMV6-XL5 (sc111136, OriGene, Rockville, MD, USA).
NAMPT promoter activity
NAMPT promoter activity in human lung EC was studied using the NAMPT promoter construct as we have previously described. 12 The Promega Dual Luciferase assay (Promega) was followed according to the protocol to assess NAMPT promoter activity in sham vector control and NAMPT SNP-transfected EC. Six (n = 6) duplicate luciferase assays were performed per condition and per the manufacturer’s instructions and as previously described. 12 Luminescence [relative light unit (RLU)] ratios were normalized to log2-untreated control (pGL3 with rs61330082 G).
Immunoblotting assays
NAMPT protein expression in human lung EC was studied as we have previously described.25,32,33 Cultured ECs were lysed in RIPA buffer in the presence of protease/phosphatase inhibitors. Protein levels in supernatants were quantified using a BCA kit (Life Technologies cat. 23227). Samples were denatured by boiling, mixed with Bolt 4× LDS sample buffer with reducing agent (Life Technologies cat. B0004), loaded in Bolt 4–12% Bis-Tris gels in MOPS SDS buffer (Life Technologies cat. B0001), and transferred to a nitrocellulose membrane. Membranes were probed with the respective primary antibodies overnight at 4°C in 5% milk-TBS, followed by incubation with their respective HRP-secondary antibodies (1 h). Immunoreactivity was detected by SuperSignal West Pico Chemiluminescent Substrate (Life Technologies cat. 34080) on ChemDoc MP Imaging System (Bio-Rad Laboratories). Primary antibodies used were rabbit anti-NAMPT pAb (Bethyl, 100 µl/well, dilution 1:10,000) and mouse anti-β-actin (CAT# sc-47778; Santa Cruz Biotechnology).
Trans-endothelial electrical resistance (TER) across human lung EC
Human pulmonary artery ECs (Lonza, Walkersville, MD, USA) were grown to confluence on evaporated gold microelectrodes in polycarbonate wells as we have described previously. 34 TER measurements were performed using an electrical cell-substrate impedance sensing system (Applied Biophysics, Troy, NY, USA). TER values from each microelectrode were pooled as discrete time points and plotted versus time as the mean ± SEM.
Statistical analysis
Statistical analysis was performed in STATA 15.1 (StataCorp. LLC: Release 15, College Station, TX, USA), GraphPad Prism v 8.0.0 (GraphPad Software, San Diego, CA, USA), and R (R Core Team, 2020). For the DISCOVERY cohort, with 474 ARDS patients and 31% mortality, we estimated a 90% statistical power to identify a significant difference in eNAMPT levels between controls and ARDS patients with a type 1 error rate of 0.05. For the validation cohort of ALVEOLI patients with 50% mortality, we estimated an 80% statistical power to identify a significant difference in eNAMPT levels between controls and ARDS patients at a type 1 error rate of 0.05. Statistical power to identify a significant difference in eNAMPT levels between survivors and non-survivors was only 72% and 60% for the DISCOVERY and ALVEOLI cohorts respectively. A two-way analysis of variance (ANOVA) was used to assess the interaction of self-reported race and mortality on plasma eNAMPT levels. An adjusted linear regression model with NAMPT diplotypes and covariates was utilized. Statistical differences were measured using either an unpaired Student’s t-test or a two-way ANOVA. When the data analyzed were not distributed normally, we used the Mann–Whitney test or Kruskal–Wallis one-way ANOVA.
Results
Functional effect of NAMPT SNP G-1535A on human EC NAMPT promoter activity
Figure 1(a) depicts the genomic location of the three NAMPT promoter SNPs analyzed in this study: G-1535A (rs61330082), A-1001 C (rs9770242), and C-948A (rs59744560). The G-1535A and A-1001 C SNPs have each been previously associated with altered ARDS risk and severity.10,22,23 We subcloned the NAMPT -1535A SNP or the wild-type allele, -1535G, into NAMPT expression plasmids encoding the NAMPT promoter luciferase reporter followed by transfection into human lung ECs. NAMPT promoter activities were increased in both -1535G- and -1535A-transfected EC in response to LPS (p < 0.01 versus vehicle) (Figure 1(b)); however, the magnitude of LPS-induced increases in NAMPT promoter activity was significantly reduced in EC harboring the -1535A variant compared with -1535G (p = 0.008) (Figure 1(b)). We observed similar significant reductions in the magnitude of LPS/18% CS (4 h)-induced NAMPT promoter activity in EC expressing the -1535A variant compared with -1535G (p < 0.001) (Figure 1(c)).
Functional effect of NAMPT -1535A/-948 C diplotype on EC NAMPT promoter activity, eNAMPT secretion, and barrier regulation
EC transfected with either -1535G/-948A or -1535A/-948 C diplotypes demonstrated significant increases in NAMPT promoter luciferase reporter activities in response to LPS (4 h), 18% cyclic stretch (CS, 4 h), or LPS/18% CS challenge (8 h) (Figure 1(d)). We next demonstrated that EC harboring the -1535G/-948A diplotype exhibited significantly greater EC promoter reporter activities compared with the -1535A/-948 C diplotype in response to LPS, 18% CS alone, or the combination of LPS/18% (Figure 1(d)), findings consistent with dampening of stimulated NAMPT promoter activity. NAMPT SNP promoter study results were validated by biochemical studies showing increased NAMPT protein expression in EC overexpressing the 1535G/-948 C plasmid compared with overexpression of 1535A/-948 C, with results quantified by densitometric analysis (Figure 1(e) and (f)).
We next examined the functional effects of -1535A/-948 C and 1535G/-948A overexpression on LPS-induced EC eNAMPT secretion into the cell media (24 h). eNAMPT secretion was increased in LPS-challenged wild-type and SNP-transfected ECs; however, the magnitude of eNAMPT secretion was significantly greater in EC overexpressing the -1535G/-948A diplotype compared with the -1535A/-948 C diplotype (Figure 1(g)). Finally, we evaluated the effect of -1535A/-948 C and 1535G/-948A diplotypes on thrombin-induced EC barrier dysfunction. 34 EC overexpressing either -1535G/-948A or the -1535A/-948 C diplotype exhibited a greater magnitude of thrombin-induced TER reductions and loss of EC integrity than untransfected ECs (Figure 1(h)). However, reductions in TER after thrombin challenge were significantly greater in EC overexpressing the -1535G/-948A compared with ECs expressing the -1535A/-948 C diplotype.
Demographics and mortality in the DISCOVERY and ALVEOLI ARDS cohorts
The demographics and clinical characteristics of both ARDS DISCOVERY and ALVEOLI cohorts are detailed in Tables 1 and 2. Overall 28-day mortality was 31% in the ARDS DISCOVERY cohort and 22.5% for the entire ALVEOLI cohort (n = 531 subjects). Mortality was 50% in the restricted ALVEOLI cohort as subjects were purposefully selected to achieve a comparable ARDS survivor/non-survivor ratio. The ARDS DISCOVERY cohort did not demonstrate significant differences between survivors and non-survivors with regard to sex, age, APACHE II score, and sepsis status (Table 1). APACHE III scores in both DISCOVERY and ALVEOLI ARDS cohorts were significantly higher in non-survivors, both in African American (AAs) and in European Americans (EAs) (Table 1, Figure 2(a)).
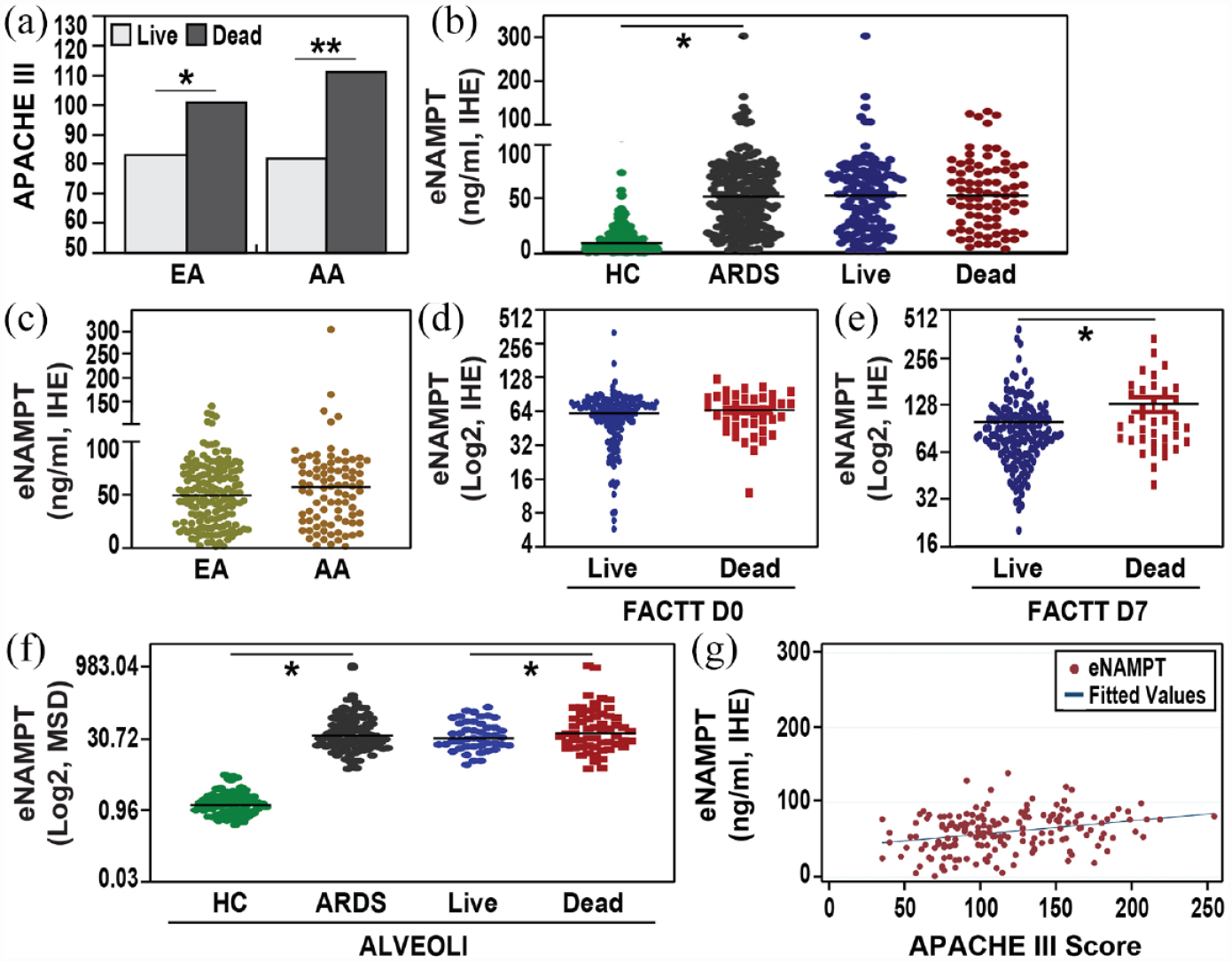
ARDS severity and plasma eNAMPT levels in ARDS DISCOVERY and ALVEOLI cohorts. (a) Shown are APACHE III scores for EAs and AAs participating in the combined ARDS DISCOVERY (n = 474) and ALVEOLI cohorts studied (n = 103) with demographics and clinical characteristics by mortality detailed in Tables 1 and 2. APACHE III scores were increased in non-ARDS survivors compared with survivors with AAs exhibiting significantly greater APACHE III scores in non-survivors compared with EAs. (b) Day 0 median plasma eNAMPT levels were significantly elevated in the ARDS DISCOVERY cohort (57.9 ± 1.75 ng/ml) using an in-house developed eNAMPT ELISA assay (IHE) compared with healthy controls (n = 268, 9.3 ± 0.9 ng/ml, p = 0001). Plasma eNAMPT values were increased in ARDS non-survivors (n = 131, 58.7 ± 0.9 ng/l), but this was not statistically significant compared with ARDS survivors (n = 297, 55.1 ± 0.9 ng/ml, p = 0.2). (c) Day 0 median plasma eNAMPT levels in EAs and AAs in the ARDS DISCOVERY cohort trended toward increases in AAs compared with EAs; however, this was not statistically significant (57 ± 41 versus 49.3 ± 28.7 ng/ml). (d) and (e) Day 0 and Day 7 plasma eNAMPT levels in ARDS FACTT DISCOVERY cohort survivors (n = 160) and non-survivors (n = 40) on Day 0 and Day 7. No significant differences in Day 0 values were observed in survivors and non-survivors (61.4 ± 6.9 ng/ml versus 64.1 ± 8.8 ng/ml, p = 0.07). In contrast, comparison of Day 7 eNAMPT plasma levels from FACTT/DISCOVERY ARDS survivors and non-survivors (n = 162 and 41, respectively) demonstrated statistically significant increases in ARDS non-survivors (90.5 ± 9.7 ng/ml versus 123.3 ± 11.1 ng/ml, p = 0.001). (f) Day 0 plasma eNAMPT levels in the ARDS ALVEOLI cohort (50% mortality, n = 103) with significantly elevated Day 0 eNAMPT levels (35.8 ± 4.3 ng/ml) compared with healthy controls (n = 78, 1.21 ± 0.9 ng/ml, p < 0.01) utilizing the MSD U-Plex assay. As shown in Table 2, median eNAMPT levels were significantly increased in non-survivors (n = 57, 39.8 ± 18.8 ng/ml) compared with survivors (n = 46, 32.3 ± 12.3 ng/ml, p = 0.04). (g) Utilizing a linear regression model, ARDS DISCOVERY cohort subjects with APACHE III severity score available, scores were significantly and directly linked to plasma eNAMPT levels (n = 157, R 2 = 0.03, adjusted R 2 = 0.02, p < 0.02).
eNAMPT plasma levels are significantly increased in ARDS DISCOVERY and ALVEOLI cohorts
Supplemental Figure 1 details the overall experimental design employed to obtain eNAMPT plasma values and NAMPT SNP genotypes in the ARDS DISCOVERY and ALVEOLI cohorts. Utilizing an in-house eNAMPT ELISA assay, the ARDS DISCOVERY cohort (n = 474) exhibited significantly elevated median eNAMPT plasma values (57.9 ± 1.75 ng/ml) compared with healthy controls (n = 268, 9.3 ± 0.9 ng/ml, p = 0001) (Figure 2(b)). Plasma eNAMPT levels trended toward increases in ARDS non-survivors (n = 131, 58.7 ± 0.9 ng/ml) compared with ARDS survivors (n = 297, 55.1 ± 0.9 ng/ml), but this was not statistically significant (p = 0.2, Figure 2(b)). Similarly, in the ARDS DISCOVERY cohort, plasma eNAMPT values were not statistically significant between EAs and AAs (p = 0.3, Figure 2(c)). Despite the trend toward higher eNAMPT levels in non-surviving AAs when compared with EAs in both D0 (54.6 versus 53.5 ng/ml) and D7 samples (77.3 versus 62.5 ng/ml), this was not statistically significant (p = 0.3 and p = 0.9 respectively). Interestingly, in contrast to many published biomarker studies consistently reporting Day 3 and Day 7 values to decline compared with Day 0 ARDS levels,35–38 ARDS FACTT DISCOVERY cohort non-survivor subjects exhibited either comparable or further increased Day 7 plasma eNAMPT levels from Day 0 values (r = 0.05, 95% CI = 0.17–0.76, p = 0.005) (Figure 2(d) and (e), Supplemental Table 2). In addition, Day 7 plasma eNAMPT levels were significantly higher in FACTT/ DISCOVERY ARDS non-survivors (n = 40, 100.9 ± 9.5 ng/ml) compared with survivors (n = 160, 83.01 ± 6.5 ng/ml, p = 0.01) (Figure 2(d) and (e)).
Similar findings were observed in the restricted ARDS ALVEOLI cohort (50% mortality, n = 103) with significantly elevated Day 0 eNAMPT levels (35.8 ± 4.3 ng/ml) compared with healthy controls (n = 78, 1.2 ± 0.9 ng/ml, p < 0.01) (MSD U-Plex assay) (Figure 2(f)). Day 0 plasma eNAMPT levels were also significantly higher in ALVEOLI ARDS non-survivors (n = 57) compared with survivors (n = 46, 39.7 ± 3.9 ng/ml versus 31.8 ± 2.3 ng/ml, p = 0.04) (Figure 2(f)). The plasma eNAMPT levels in ARDS non-survivors in DISCOVERY and ALVEOLI cohorts are presented in Supplemental Table 3. Finally, utilizing a linear regression model, ARDS DISCOVERY cohort subjects with APACHE III severity score availability (n = 209) were significantly linked to plasma eNAMPT levels (n = 157, adjusted R 2 = 0.02, p < 0.02) (Figure 2(g)). Significant differences were not observed between eNAMPT levels in sepsis versus non-sepsis subgroups in the DISCOVERY (mean = 50.9 and 53.9 ng/ml, respectively) and VALIDATION cohorts (mean = 74 and 100 ng/ml, respectively).
NAMPT SNP minor allelic frequencies (MAFs) in ARDS DISCOVERY and ALVEOLI cohorts compared with the 1000 Human Genome Project dataset
We next compared MAFs in the combined ARDS cohorts and the 1000 Human Genome Project dataset (Figure 3(a)), revealing that the -1535A SNP allele is reduced in both ARDS EAs (p = 0.07) and AAs (p = 0.0006) compared with 1000 Human Genome subjects (Figure 3(a)). In contrast, the MAF for the -1001 C SNP is increased in both EA and AA ARDS cohort subjects although it was not significantly different when compared with 1000 Human Genome Project subjects (Figure 3(a)). Finally, the C-948A SNP MAF was more common in ARDS EAs (p = 0.11) and significantly more common in ARDS AAs (p = 0.00001) compared with 1000 Human Genome Project subjects (Figure 3(a)). Unfortunately, comparisons of MAFs within the combined DISCOVERY and ALVEOLI ARDS cohorts (n = 508) failed to demonstrate significant NAMPT SNP association with either APACHE III scores or plasma eNAMPT comparisons (Figure 3(b)–(d)), findings we speculate to reflect the lack of sufficient statistical power for these rigorous statistical correlations.

Allelic association of NAMPT promoter SNPs with ARDS risk and plasma eNAMPT levels in ARDS DISCOVERY subjects. (a) NAMPT SNP minor allelic frequency (MAF) in ARDS DISCOVERY and ALVEOLI cohorts (light gray bars) compared with the 1000 Human Genome Project dataset (dark gray bars). The -1535A SNP allele is reduced in ARDS EAs (p = 0.07) and AAs (p = 0.0006) compared with 1000 Human Genome subjects. The MAF for the -1001 C SNP was not significantly different between EAs and AAs in the ARDS cohorts and the 1000 Human Genome Project subjects. The C-948A SNP frequency was greater in ARDS EAs (p = 0.11) and ARDS AAs (p = 0.00001). (b–d) Comparisons of MAFs for G-1535A, T-1001 C, and C-948A against the APACHE III scores within the combined DISCOVERY and ALVEOLI ARDS cohorts (n = 508) failed to demonstrate significant SNP association with either APACHE III scores (light gray bars) or plasma eNAMPT comparisons (dark gray bars).
Comparison of NAMPT SNP diplotype frequencies in ARDS DISCOVERY and ALVEOLI cohorts and the 1000 Human Genome Project dataset
Finally, similar to Figure 3(a), we next compared G-1535A, T-1001 C, and C-948A diplotype frequencies for EAs and AAs in the combined ARDS cohorts and the 1000 Human Genome Project (Figure 4). Figure 4(a) and (b) demonstrates that the frequency of the risk diplotype -1535G/-948A diplotype (Figure 4(b)) in EAs and AAs in the combined ARDS cohorts was significantly more frequent compared with the 1000 Human Genome Project. Conversely, the protective -1535A/-948 C diplotype was significantly less frequent in both EAs and AAs in the combined ARDS cohorts with the protective diplotype much less common in AAs than EAs. Similarly, Figure 4(c) and (d) demonstrates that the frequency of the risk diplotype -1535G/-1001CA diplotype (Figure 4(b)) and the protective -1535A/-1001A diplotype significantly differed in EAs and AAs in the combined ARDS cohorts compared with the 1000 Human Genome Project, again with the protective -1535A/-1001A diplotype far less common in AAs.
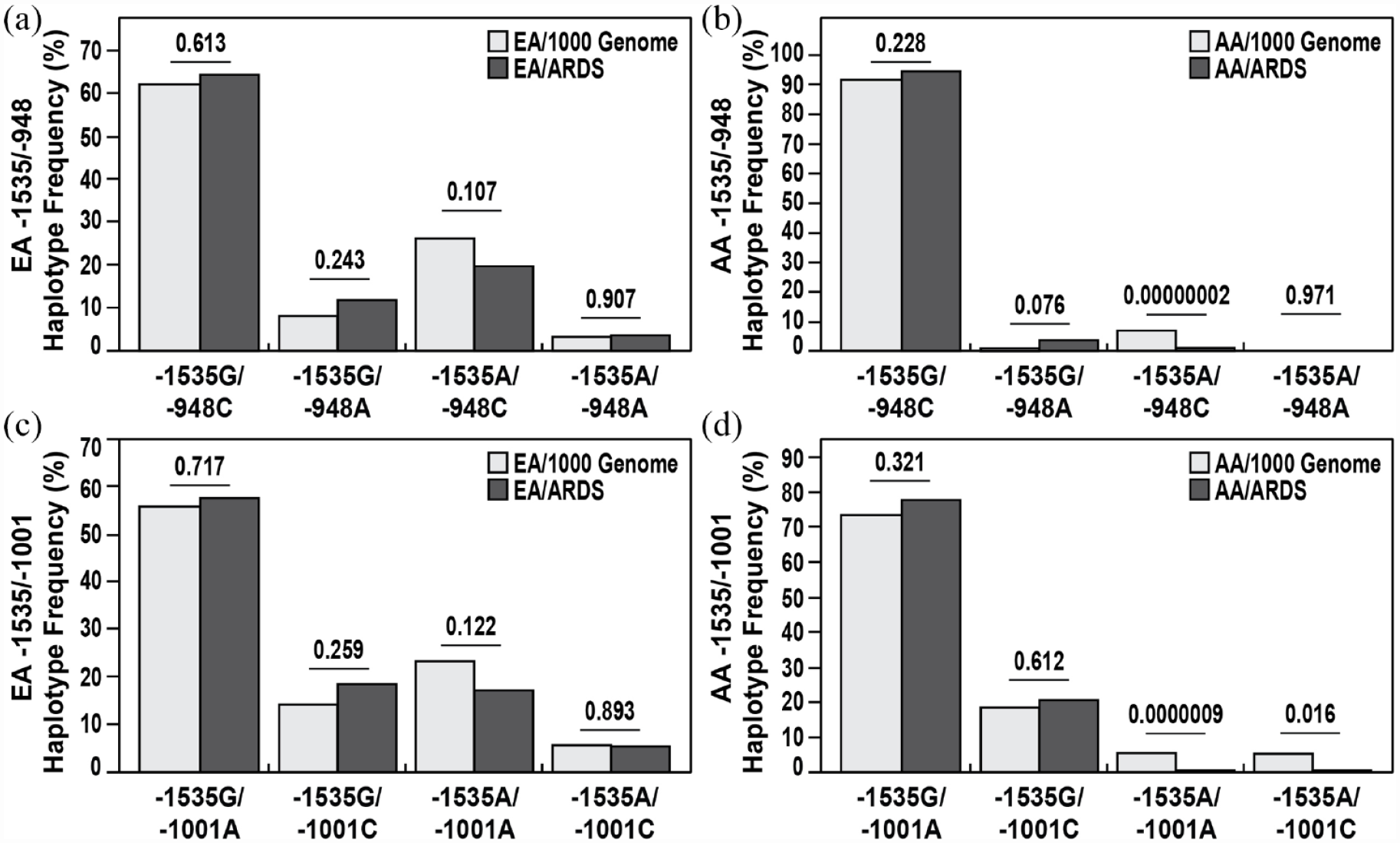
Comparison of NAMPT SNP diplotype frequencies in combined ARDS DISCOVERY and ALVEOLI cohorts and the 1000 Human Genome Project dataset. (a) and (b) G-1535A and C-948A diplotype frequencies for EAs and AAs were compared in the combined ARDS cohorts and the 1000 Human Genome Project. The risk diplotype -1535G/-948A diplotype was significantly increased in the combined ARDS cohorts compared with the 1000 Human Genome Project, both in EAs and in AAs. Conversely, the protective -1535A/-948 C diplotype was significantly reduced in both EAs and AAs in the combined ARDS cohorts with the protective diplotype much less common in AAs than in EAs. (c) and (d) The frequency of the risk diplotype -1535G/-1001CA diplotype and the protective -1535A/-1001A diplotype significantly differed in EAs and AAs in the combined ARDS cohorts compared with the 1000 Human Genome Project, again with the protective -1535A/-1001A diplotype far less common in AAs.
Discussion
ARDS is a vexing critical illness made more challenging by the lack of FDA-approved therapies and the sobering failure of therapeutically-driven ARDS clinical trials.39,40 Contributing to the absence of successful phase II/III trials is the inability to stratify ARDS patients, either by mortality risk or by the likelihood of drug responsiveness, and the absence of useful predictive biomarkers or genetic variants in the critically ill.17,41 Although an inflammatory subphenotype among multiple ARDS cohorts has been recently described as significantly associated with sepsis outcomes and ARDS mortality, 39 a validated prognostic biomarker that reliably identifies this ARDS inflammatory subphenotype has not been identified or successfully utilized in clinical trials. ARDS subject stratification by any modality would have significant utility in the clinical management of ARDS patients and in clinical trial design for novel therapeutics.17,28,39,42
Genomic and genetic approaches to deciphering ARDS risk and mortality have been reported.43,44 Our prior systems biology and translational approach to ARDS led to the identification of NAMPT as an ARDS candidate gene with ARDS-associated variants,10–12 which was further confirmed in other reports,22,23 and eNAMPT as a potentially useful ICU biomarker 17,18,21 and viable therapeutic target in ARDS/VILI.13–16,27 The present report extends our prior clinical and preclinical work to address potential mechanistic insights into the association of NAMPT SNPs on ARDS severity. We performed in vitro studies in human lung ECs, a key ARDS target tissue, 27 transfected with NAMPT promoter luciferase reporters harboring wild-type or NAMPT SNPs (G-1535A, T-1001 C, C-948A), as well as in vivo studies utilizing two well-phenotyped human ARDS cohorts. Predictably, the two human ARDS cohorts exhibited APACHE III scores which were higher in EA and AA non-survivors compared with EA and AA survivors, with AA non-survivors exhibiting greater APACHE III scores than EA non-survivors (Figure 2). In addition, eNAMPT plasma levels were significantly increased in both ARDS cohorts compared with controls and were linked to ARDS severity (Figure 2).
The mechanistic influence of NAMPT promoter SNPs on plasma eNAMPT levels was demonstrated by the protective nature of the -1535A/-948 C haplotype in dampening inflammation-induced increases in NAMPT promoter activity, NAMPT protein expression, eNAMPT secretion, and vascular permeability (Figure 1), findings confirming the previously reported significant association of the -1535A SNP with lower ARDS severity.10,22,23 When the frequency of this protective diplotype was compared between ARDS subjects and the 1000 Human Genome Project healthy subjects, there was a significant reduction in diplotype frequency in ARDS subjects (Figure 4). In contrast, the -1535G/-948A diplotype was found to significantly increase NAMPT promoter activity, NAMPT protein expression, eNAMPT secretion, and the magnitude of thrombin-induced vascular permeability when compared with -1535A/-948 C, findings corroborated by comparisons between ARDS subjects and the 1000 Human Genome Project healthy subjects (Figure 4). Unfortunately, although MAFs for -1001 C were greater in both EAs and AAs in the ARDS DISCOVERY and ALVEOLI cohorts, this did not achieve statistical significance when compared with the 1000 Human Genome Project healthy subjects (Figure 4). In addition, similar to -1535A and -948A, the -1001 C SNP was not significantly associated either with plasma eNAMPT levels or with APACHE III scores in our two ARDS cohorts. We speculate these results to reflect the lack of sufficient statistical power to allow for these rigorous statistical correlations.
An important aspect of our study is the inclusion of a sizable AA ARDS population in the ARDS DISCOVERY cohort, an ‘at-risk’ population with increasingly recognized disparities in both ARDS incidence and severity,45,46 especially in the current landscape of the COVID-19 pandemic. 2 Several ARDS genes, including MYLK (myosin light chain kinase),47,48 MIF (macrophage migration inhibitory factor), 7 GADD45a (growth arrest DNA damage inducible alpha), 9 S1PR3 (sphingosine-1-phosphate receptor-3), 49 and SELPLG (selectin P ligand gene), 6 have previously been shown to harbor SNPs significantly associated with the development of ARDS in AA populations. We identified significant reductions in MAFs for the -1535A SNP (p = 0.0006) and the C-948A SNP as in AAs (p = 0.00001) compared with AAs in the 1000 Human Genome Project (Figure 3(a)). Unfortunately, comparisons of MAFs in AAs within the combined DISCOVERY and ALVEOLI ARDS cohorts (n = 508) failed to demonstrate significant SNP association with either APACHE III scores or plasma eNAMPT comparisons (Figure 3(b)–(d)), findings we again speculate to reflect the lack of sufficient statistical power for these rigorous statistical correlations. Corroborating these findings for -1535A SNP and the C-948A SNP in AAs, comparison of NAMPT SNP risk -1535G/-948A diplotype in AAs in the combined ARDS cohorts was significantly increased compared with the 1000 Human Genome Project and the protective -1535A/-948 C diplotype was significantly reduced in AAs. In the combined ARDS cohorts, the protective diplotype is significantly less common in AAs versus EAs (Figure 4(b)), a feature shared by the assessment of the protective -1535A/-1001A diplotype in AAs (Figure 4(b)).
We readily recognize several important limitations in our genotype–phenotype-driven studies. Foremost of study limitations is the absence of a large, standardized cohort to validate the potential clinical utility of NAMPT SNPs and eNAMPT plasma levels to be of potential utility in ARDS Clinical Trial design, as depicted in Figure 5. In addition, we recognize additional important limitations including the heterogeneity attributable to the various cohorts included in our DISCOVERY set, the limited sample availability, missing mortality data hampering full assessment of the prognostic capacity of eNAMPT levels on the risk of death, or discharge prior to Day 7, that is, sample selection bias. These factors limited our capacity to expand our analysis to study the effect of MIF in subphenotypic groups by race and mortality. APACHE scores reduced the availability of plasma eNAMPT determinations across the ARDS DISCOVERY cohort entire cohort, a finding which further limited the power of our genotype–phenotype association analyses. To increase the predictive power of our hypothesized clinical trial stratification strategy requires a future validation study in a larger replication cohort that may allow for the addition of additional risk NAMPT SNPs for clinical trial risk stratification. The correlations between other haplotypes, including -948/-1001, and NAMPT promoter activity and protein secretion in ARDS models also require further investigation. Nevertheless, our results are consistent with prior reported effects of ARDS-associated protective SNPs and risk SNPs on NAMPT promoter activity and mRNA expression.10,12,24

A hypothesized precision medicine strategy for ARDS clinical trial subject stratification. Shown is a schematic diagram delineating the potential utility of NAMPT genotypes for -1535, -1001, and -948 and Day 0 eNAMPT plasma levels in ARDS subject recruitment to clinical trials. Individuals with respiratory distress and severe hypoxemia are evaluated for eNAMPT plasma levels and NAMPT genotypes. Individuals with elevated eNAMPT plasma levels and high-risk NAMPT genotypes would be considered high-risk ARDS subjects and an optimal cohort for potential recruitment to an ARDS therapeutic trial for evaluation of responses to a mortality-sparing, eNAMPT-focused therapeutic mAb. Individuals exhibiting eNAMPT plasma levels <10 ng/ml and low-risk NAMPT genotypes would be considered ineligible for recruitment.
In summary, we explored the utility of integrating plasma levels of eNAMPT, a highly druggable ARDS therapeutic target, with NAMPT genotypes as a potential approach to the unmet need for improved therapeutic clinical trial design in ARDS. We defined the mechanistic influence of NAMPT promoter SNPs (G-1535A, T-1001 C, C-948A) in human lung ECs, a key ARDS target tissue, in regulating NAMPT promoter activity, NAMPT protein expression, and eNAMPT secretion. The current work is the first to establish a plausible genotype–phenotype linkage between -1535/-948 NAMPT diplotypes, eNAMPT plasma levels in ARDS patients, and ARDS severity utilizing two human ARDS cohorts (n = >500). eNAMPT plasma levels and the risk NAMPT genotype represent novel stratification tools to increase the success of eNAMPT-focused therapeutic clinical trials in high-risk ARDS subjects.
Supplemental Material
sj-docx-2-tar-10.1177_17534666231181262 – Supplemental material for Linkage of NAMPT promoter variants to eNAMPT secretion, plasma eNAMPT levels, and ARDS severity
Supplemental material, sj-docx-2-tar-10.1177_17534666231181262 for Linkage of NAMPT promoter variants to eNAMPT secretion, plasma eNAMPT levels, and ARDS severity by Heather Lynn, Xiaoguang Sun, Nancy G. Casanova, Christian Bime, Vivian Reyes Hernon, Clayton Lanham, Radu C. Oita, Nikolas Ramos, Belinda Sun, Dawn K. Coletta, Sara M. Camp, Jason H. Karnes, Nathan A. Ellis and Joe G.N. Garcia in Therapeutic Advances in Respiratory Disease
Supplemental Material
sj-png-1-tar-10.1177_17534666231181262 – Supplemental material for Linkage of NAMPT promoter variants to eNAMPT secretion, plasma eNAMPT levels, and ARDS severity
Supplemental material, sj-png-1-tar-10.1177_17534666231181262 for Linkage of NAMPT promoter variants to eNAMPT secretion, plasma eNAMPT levels, and ARDS severity by Heather Lynn, Xiaoguang Sun, Nancy G. Casanova, Christian Bime, Vivian Reyes Hernon, Clayton Lanham, Radu C. Oita, Nikolas Ramos, Belinda Sun, Dawn K. Coletta, Sara M. Camp, Jason H. Karnes, Nathan A. Ellis and Joe G.N. Garcia in Therapeutic Advances in Respiratory Disease
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.