
Abstract
Coronavirus-induced diseases have afflicted humanity for several decades. This scenario was aggravated by the emergence of the coronavirus disease 2019 (named COVID-19) in Wuhan, China, in December 2019. Since then, COVID-19 has killed millions of people worldwide, probably the most devastating pandemic since HIV/AIDS. This review aimed to bring together important updated aspects related to coronavirus-induced diseases and the enhanced vascular permeability observed mainly in the lungs of affected people. The dysregulated vascular permeability in the lungs is of fundamental importance for coronaviruses-caused morbidity and mortality. Thus, as described in this review, it is a target of new and old drugs.
Introduction
Since the discovery of the first virus – the tobacco mosaic virus – at the end of the 19th Century, a possible involvement of those intracellular obligatory-living creatures with human diseases was proposed. Today, it is known that more than 200 viruses can infect humans, with the first of them, the yellow fever virus, being discovered in 1901. 1 After the great panic caused by the human immunodeficiency virus 1 (HIV-1) in the world population about 40 years ago, another virus has recently terrorized humans, the severe acute respiratory syndrome coronavirus 2 virus (SARS-CoV-2). The scary feeling related to coronavirus disease 2019 (COVID-19) – the disease caused by the SARS-CoV-2 – comes from the impressive numbers observed in those infected and not previously vaccinated: about 14–20% progressed to severe disease, 5% became critically ill, and 2% died.2,3 Besides, survivors may present late symptoms, commonly including fatigue, dyspnea, and cognitive impairment, months after the onset of acute symptoms. 4 For those with severe COVID-19, a ‘cytokine storm’ is observed, along with profound liquid accumulation in the lungs, which turns breathing into an agonizing task. In this review, we will bring the most recent aspects related to the coronavirus infection and the enhanced vascular permeability that happens in the lungs of infected people with mild to severe symptoms.
Furthermore, we point out some molecular targets that new drugs or existing ones (repurposing) can act to reduce lung edema and, consequently, intensive care unit (ICU) admission and mortality. Initially, general aspects of the coronavirus family will be discussed, followed by a series of mechanisms coronavirus uses to enhance vascular permeability and potential targets to reduce blood vessel leakage in the lungs. Later, we discuss some drugs that have been shown to diminish lung edema.
Coronaviruses and diseases
Coronaviruses make part of a family of viruses called Coronaviridae. They are enveloped viruses with a single-strand positive-sense RNA and a crown-like morphology that gives their family’s name.5,6 There are four subfamilies: alphacoronavirus, betacoronavirus, gammacoronavirus, and deltacoronavirus, with distinction observed based on their phylogenetic relationship and genomic structure. 7 Alphacoronaviruses and betacoronaviruses infect only mammals; gammacoronaviruses and deltacoronaviruses infect birds.7,8
Human coronaviruses (HCoVs) like HCoV-HKU1, HCoV-NL63, HCoV-229E, and HCoV-OC43 usually cause mild respiratory disease and affect mainly the upper respiratory tract with symptoms like cough, sore throat, and coryza. On the contrary, other coronaviruses that induce lower respiratory tract diseases have afflicted people worldwide in the last decades. In fact, in 2002–2003, there was an outbreak of the severe acute respiratory syndrome coronavirus (SARS-CoV) with thousands of cases and several hundred deaths in 30 countries.9–11 Similar numbers were registered one decade later with the outbreak of the Middle East respiratory coronavirus (MERS-CoV).9,11 Lately, with the first case registered in December 2019, a novel betacoronavirus named SARS-CoV-2 is responsible for the global outbreak of a respiratory illness called COVID-19. According to the World Health Organization (WHO), COVID-19 has been confirmed in almost 500 million people, and more than 6 million deaths have been documented worldwide. Despite a dramatic reduction in the number of infected and deaths reported after some vaccines were developed, COVID-19 cases and deaths are continuously registered in expressive numbers worldwide every day.
Coronaviruses traverse across species leading to humans. Animals suspected to host some of these viruses include Himalayan palm civets (Paguma larvata), raccoon dogs (Nyctereutes procyonoides), dromedary camels (Camelus dromedarius), and bats (Rhinolophidae and Neoromicia capensis). This hypothesis accompanied the SARS-CoV, MERS-CoV, and SARS-CoV-2 infections.12–18
SARS-CoV and MERS-CoV infections usually result in severe pneumonia. 11 SARS-CoV utilizes a receptor called angiotensin-converting enzyme 2 (ACE2R) placed on the external membrane of some cells – ciliated bronchial epithelial cells, types I and II epithelial cells, endothelium – in the host to infect humans.19,20 ACE2R expression is found mainly in the lungs and other organs like the heart, kidney, and intestine.21,22 Another protein in the host named CD209L (a C-type lectin, also called L-SIGN) is expressed in type II alveolar cells and endothelial cells and was shown to be necessary for virus internalization as well. 23 A viral spike (S) protein binds the ACE2R and CD209L and promotes virus entrance.23,24 Furthermore, a protein called 3CLpro, which originated after the lysis of other proteins, particularly pp1a and pp1ab, plays a vital role in the SARS-CoV life cycle and is another potential target for drugs. 25 Other structural proteins essential to virus assembly include envelope (E), matrix (M), and nucleocapsid (N) (Figure 1). 26

SARS-CoV structural proteins S, N, M, and E, and pp1a and pp1b.
Fever, myalgia, malaise, and cough are common initial symptoms observed in many patients infected with SARS-CoV.27,28 A high-resolution computed tomography (CT) unravels ground-grass opacifications as a typical initial abnormality in the lungs of about 67% of patients. 29 One-third of patients get better after a few days; the others commonly evolve to diffuse alveolar damage, which usually and rapidly progresses to shortness of breath and respiratory distress, leading to an estimated average of 20–30% of patients needing intensive care, and about 10% of them dying.27–32 Ventilatory assistance is often required in patients with respiratory distress. Lung histology is characterized by epithelial cell hyperplasia and detachment, hyalinization, edema, inflammatory cell infiltration (monocytes, macrophages, lymphocytes, and neutrophils), and evolution to fibrosis.33–35 Furthermore, a SARS viral protein – 7a – can induce apoptosis in cells and directly contribute to tissue damage. 36
The pathophysiology of the SARS-CoV infection also involves indirect mechanisms, mainly through the overactivation of the immune system. Initially, dendritic cells and alveolar macrophages phagocyte virus-infected epithelial type 2 alveolar cells, resulting in a massive neutrophil attraction and T-cell activation. 22 Moreover, a profound increase in cytokine production, mainly interleukin 6 (IL)-6, IL-8, interferon-inducible protein 10 [IP-10 or CXC-motif chemokines ligand (CXCL10)], and tumor necrosis factor-alpha (TNF-α), the so-called ‘cytokine storm’, along with a gradation of a reduced number of lymphocytes and enhanced neutrophils, which is thought to contribute to the mortality index (10%) significantly.37,38 The absence of a potent antivirus T-cell response could lead to a dysregulated innate response and poor outcomes. 39 It is not known whether SARS-CoV infects T-cells. Mechanisms to explain T-cell reducing number include altered antigen-presenting cell (APC) function, impaired dendritic cell migration to lymph nodes, pronounced interferon-gamma (IFN-γ) expression, or glucocorticoid responses, all of which can induce T-cell apoptosis.40–42 Corroborating this last hypothesis, not only IFN-γ but also other circulating cytokine levels were also reported to be enhanced in SARS-CoV-infected patients, including IL-1, IL-8, IL-12, IL-18, monocyte chemotactic protein 1 (MCP1 or CCL2), monokine induced by IFN-γ (MIG), and transforming growth factor-β (TGF-β), characterizing a Th1 inflammatory response.43–45 Besides the acute effects, post-traumatic changes after SARS-CoV infection were reported in many patients months after hospital discharge. 46
Independent prognostic factors for the worst outcome in SARS-CoV-infected patients include advanced age, diabetes mellitus, and heart disease.47,48 Different treatments were utilized, including ribavirin, lopinavir/ritonavir, or corticosteroids, with the latter yielding better results. Large randomized clinical trials are lacking, however, and no commercially available vaccine prevents SARS infection.27,49–53
MERS-CoV is constituted of four structural proteins similar to SARS-CoV: S, M, E, and N. Main common symptoms include fever, cough, sore throat, shortness of breath, and myalgia. Radiographs and CT showed subtle or extensive unilateral or bilateral changes with ground-glass opacities and limited consolidation.54,55 Although no report describing the pathophysiological changes in the lungs of patients who died due to MERS-CoV infection has been published, tissues infected ex vivo exhibited epithelial cell detachment, tight junction disruption, and cell apoptosis. 56 These results may explain why many patients commonly progress to respiratory distress, need respiratory assistance (mechanical ventilation), and eventually die. As observed in SARS infection, old age, diabetes mellitus, and chronic renal disease are risk factors for the worst prognosis in patients with MERS-CoV. 57 On the contrary, some differences exist between SARS and MERS: MERS-CoV uses the dipeptidyl peptidase 4 (DPP4) (also called CD26) receptor to enter the host cells instead of ACE2, as seen with SARS-CoV,58,59 human-to-human disease spread is not so efficient as that seen in SARS infection,60–62 and the mortality rate is elevated by 30% compared with SARS (10%). 63 DPP4 is expressed in various cells, including type I and II alveolar epithelial cells, ciliated and non-ciliated bronchial epithelium and submucosal glands, endothelium, alveolar macrophages, and leukocytes. 64
As mentioned above, studies on the pathophysiology of MERS infection are scarce. It is known that MERS-CoV downregulates genes involved in the antigen presentation pathway, including both type I and II major histocompatibility complex (MHC) genes, impairing the adaptative immune response. 65 Limited data on the cytokine profile of patients infected with MERS-CoV are available. One study showed that in bronchoalveolar lavage (BAL) and serum of a patient who died after MERS-CoV infection, there was decreased IFN-α, retinoic inducible-acid gene (RIG)-1, melanoma differentiation–associated protein 5 (MDA5), and interferon regulatory factors (IRF)3 and IRF7, which usually recognize virus invasion in the host. Furthermore, it was demonstrated high circulating levels of CXCL10 and IL-10. High levels of these mediators may lead to lower IFN-γ expression and higher levels of IL-17A and IL-23. IFN-γ is vital to combat virus infections, and high IL-17A and IL-23 often result in tissue damage. High levels of IL-12 and IFN-γ were found in a patient who survived the MERS-CoV infection. 66 Concerning the circulating immune cells number, a lesser degree of leukopenia (14% of patients) and lymphopenia (34% of patients) was observed in patients with MERS compared with SARS (47% and above 80%, respectively).67,68 The mechanism to explain MERS-CoV-induced lymphopenia remains elusive, however.
Like the SARS-CoV infection, the SARS-CoV-2 infection compromises several organs, including the heart, gastrointestinal tract, liver, brain, and kidneys, but pneumonia is the main finding. Acute respiratory failure is observed in about 14–20% of the unvaccinated SARS-CoV-2-infected symptomatic patients.2,3,69 SARS-CoV-2 infects mainly epithelial cells of pharyngeal mucosa, alveolar cells, and distal bronchial cells, but also intestinal epithelium, renal tubular epithelium, myocardial interstitial cells, cardiomyocytes, lymphoid tissue, endothelium, and pericytes.70,71 Thus, the initial most common symptoms of COVID-19 are cough, fever, fatigue, headache, myalgias, and diarrhea, and in many patients, rapid progression to dyspnea and respiratory failure occur.72,73 Other similarities between SARS-CoV and SARS-CoV-2 viruses include the structural proteins N, M, S, and E, the S protein being used to infect hosts by binding the ACE2R, patchy bilateral shadows or ground-glass opacity in lungs of most infected patients, and a worse outcome often observed in patients with comorbidities, including hypertension, diabetes and obesity, and older adults.73–78
The pathophysiology of the SARS-CoV-2 infection involves entry into cells, mainly the type II pneumocytes, by binding ACE2R and coreceptors, among them, the protease transmembrane protease serine 2 (TMPRSS-2) and cathepsin L, which cleave the S protein on the viral particle to permit engagement with ACE2R.2,79,80 This results in resident cell – for example, epithelial cells, endothelium, and alveolar macrophages – and immune cell – for example, neutrophils, CD4+ T-helper and CD8+ T-helper cells, and B cells – activation and ACE2R entrance into the epithelial cells along with the SARS-CoV-2 virus.70,81,82 Consequently, complement activation, leukocyte recruiting, antigen presentation, and additional cytokine, chemokine, enzyme, and reactive oxygen species (ROS) release are observed, ultimately injuring resident cells and tissue.77,83,84
The angiotensin (Ang) 1–7/ACE2R axis activation usually counteracts the angiotensin II (Ang II)/AT1R axis. Once activated, AT1R leads to increased production of inflammatory cytokines, exerts pro-thrombotic effects, causes vasoconstriction, releases aldosterone, and enhances ROS production, culminating in endothelial cell dysfunction and fibrosis. Thus, the dysbalanced Ang1-7/ACE2R axis may result in the overactivation of the AngII/AT1R axis.85,86 In mild to severe COVID-19 patients, profound increases in cytokine production, including IL-1β, IL-6, IL-2R, TNF-α, and IFN-γ (‘cytokine storm’), fibrosis, and microthrombi formation, including thrombotic microangiopathy in patients with severe disease, are usually observed.70,75,87–89 Thus, besides immune and resident cells overreaction, unbalanced AT1R signaling in endothelium and vascular smooth muscles may contribute to the cytokine storm, fibrosis, and microthrombi formation observed in several patients infected with SARS-CoV-2.84,85,88 Microthrombi formation was even considered a marker of COVID-19 severity, and other mechanisms may favor it, including (1) direct injury of endothelial cells by the virus with activation of the coagulation cascade, (2) neutrophil-derived tissue factor (TF)-rich neutrophil extracellular traps (NETs) formation, favoring platelet clot and activation of the extracellular pathway of coagulation, (3) enhanced neutrophil and platelet adhesion to microvessels, leading to hypoxia and upregulation of the TF expression, (4) activation of complement factors, and (5) high numbers of both thrombospondin 1 (THBS-1)+ monocytes and Myl9+ platelets.90–93 THBS-1 and Myl9 were shown to contribute to thrombus formation. THBS-1 controls platelet–endothelial cells interaction and the von Willebrand factor-dependent thrombus formation,94,95 and Myl9 was reported to form the net-like structure in the blood vessels and binds CD69-expressing leukocytes, favoring the leukocyte and platelet interaction. 96
As mentioned, thrombotic microangiopathy is often reported in COVID-19 patients with severe disease. 89 It is characterized by widespread thrombosis in capillaries and arterioles clinically manifested by microangiopathic hemolytic anemia, thrombocytopenia, and organ damage. 89 Acute kidney disease may accompany thrombotic microangiopathy in some COVID-19 patients. 97 Activated platelets and NETs play an essential role in full thrombotic microangiopathy development. 89 Furthermore, in SARS-CoV-2 infection, thrombotic microangiopathy appears triggered in some individuals by uncontrolled complement activation and genetic background. 89 Additional studies are needed to elucidate how the SARS-CoV-2 virus and thrombotic microangiopathy interact.
A reduced number of lymphocytes and prevalence of neutrophils are also commonly observed in advanced or severe COVID-19 and were associated with high mortality index (2–4%).78,98–100 A mechanism of lymphocyte reduction is still being investigated. It, however, seems to involve massive levels of TNF-α and IL-6, which leads to lymphocyte apoptosis or a direct attack on lymphocytes or lymphatic organs by the virus. 101
Higher neutrophil prevalence in an inflammatory context implies disseminated NETs release. NETs are formed by decondensed chromatin, histones, and antimicrobial proteins and function as a unique tool for killing 102 and trapping microbes, preventing them from invading tissues and organs freely.103,104 In the context of COVID-19, an increased presence of NETs in plasma and lungs was reported. As mentioned above, they contribute to thrombi formation and thrombotic microangiopathy in COVID-19 patients. 92 Besides, NET-induced endothelial cell injury was suggested. 105
All these biomolecular changes observed in COVID-19 patients with severe disease contribute to the fatal trajectory (2–4% of infected); it usually ends up with lung collapse due to alveolar-capillary barrier failure, tissue ischemia, and fibrosis.106,107
Some survivors (10–30%) may present late symptoms months after the onset of COVID-19 acute symptoms, called long COVID-19 or postacute sequelae of COVID-19 (PASC). 4 The most common symptoms of long COVID-19 are fatigue, dyspnea, and cognitive impairments. Molecular determinants of long COVID-19 seem to involve different biological systems, including the immune, endocrine, and neural systems. 4 Chronic consequences of acute dysfunctions also play a role in the long COVID-19. Indeed, fibrosis was detected in COVID-19 patients’ lung tissue months after hospital discharge. 108 Drugs to prevent long COVID-19 are still missing, however.
Similarities and differences among SARS-CoV-, MERS-CoV-, and SARS-CoV-2-induced infection in humans can be found in Table 1.
Similarities and differences among coronavirus-induced diseases.

ACE2R, angiotensin-converting enzyme 2 receptor; DPP4, dipeptidyl peptidase 4; MERS-CoV, Middle East respiratory coronavirus; SARS-CoV, severe acute respiratory syndrome coronavirus; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2 virus; TMPRSS2, transmembrane protease serine 2.
Blood vessel permeability
A significant part of blood vessels in the body is constituted of three layers – intima, media and adventitia – and are responsible for controlling blood flow to tissues and organs, supplying them with nutrients and oxygen, and clearing metabolic waste products. 110 Furthermore, blood vessels play a critical role during inflammation as their permeability is changed by inflammatory stimuli, allowing the passage of substantial amounts of fluid, large plasma proteins, including immunoglobulins, and immune cells to the injury site to combat an aggressive event.
Endothelial cells are located in the inner cellular lining of blood vessels, that is, placed in the intima layer, and are the primary regulators of permeability during inflammation. 111 Besides, even in the absence of inflammation, a different disposition of endothelial cells in the organs can make it harder or facilitate the passage of substances from the blood to tissues and vice versa; for instance, endothelial cells from cerebral blood vessels are tightly connected in a way to protect the cerebral parenchyma from plasma leakage (edema) or circulating agents. These endothelial cells, pericytes, and glial cells form the blood–brain barrier (BBB). 112 On the contrary, organs like the liver and endocrine glands keep endothelial cells relatively distant from each other, forming a structure termed gap or fenestrae that allows filtration and release of substances into circulation. 113 These fenestrae can still differ by the presence of a nonmembranous thin (5–10 ηm in thickness) structure named the diaphragm. 114 The absence of the diaphragm allows free passage of macromolecules, as seen in the liver, and its presence restricts solute passage.
In resting conditions, most organs, blood vessels, mainly capillaries, allow passage of small solutes (smaller than 40 KDa and 3 ηm in diameter) but not of larger molecules and cells as the endothelial cell line is usually uninterrupted. 115 This sieving function of blood vessels keeps the interstitial pressure and helps the immune system recognize intruders as small molecules are presented to immune cells in the lymph nodes after leaving blood vessels and entering lymphatic vessels. 116 Conversely, endothelial cells suffer molecular changes following aggressive stimuli that allow the passage of larger-sized substances and cells, mainly through postcapillary venules. 117 Permeability increases in the following order in the vasculature: arterioles, capillaries and, as the most permeable, venules.
Mechanisms underlying vascular leakage vary according to the organ, nature of what is transported – molecules or cells – and the presence of inflammatory stimuli mainly into two types: transport through vesicles or vacuoles – termed transcellular permeability – which forms a vesiculo-vacuolar organelle (VVO) after inflammatory stimuli that allows passage of macromolecules and cells, or after endothelial junctions reorganization – called paracellular permeability – which usually allows passage of fluids and small solutes in resting conditions, or even large solutes when an inflammatory stimulus is present (Figure 2).118–120 Calveolae-mediated transport of macromolecules through endothelial cells also seems to play a role in specific organs. 121

Transcellular and paracellular blood vessel permeabilities. In (a), endothelial cell vesiculo-vacuolar organelle-mediated transcellular permeability of cells and macromolecules to the parenchyma. In (b), the paracellular permeability of small solutes, fluids, and large solutes between endothelial cells in healthy and inflamed tissues.
The VVO transport occurs through the venular endothelium, and its mechanism remains unknown. Before, they believed caveolae were an essential component of the VVO structure; however, Chang et al. 122 detected VVO in the vasculature of caveolin-1 knock-out mice, reducing the importance of caveolin-1, the main protein-forming caveolae, for VVO formation.
Endothelial junctions are organized into adherens and tight junctions, also known as the zona adherens and zona occludens. 111 While tight junctions are mainly placed in the upper part of the endothelial cleft, adherens junctions go deeper into the cleft. The main component of adherens junctions is the vascular endothelial (VE)-cadherin (also termed CD144), a transmembrane protein that forms homophilic complexes between endothelial cells. 123 Adherens junctions seem to lose their function in response to several stimuli, including vascular endothelial growth factors (VEGFs), thrombin, histamine, serotonin, bradykinin, and substance P, enhancing extravasation of fluid and small solutes and diffusion of macromolecules between neighboring endothelial cells.111,124,125 In response to these stimuli, enhanced calcium entrance into the cytoplasm is observed, followed by complex formation with calmodulin and interaction with actin and myosin fibers, leading to endothelial cell contraction – that is, turns them from a flattened to a rounded shape – and dissolution of the VE-cadherin binding complex. 126 Besides, stimuli-mediated phosphorylation of VE-cadherin is pivotal to VE-cadherin internalization. Enhanced blood flow can also lead to VE-cadherin phosphorylation and results in macromolecule leakage. 127 Loss of function of the adherens junction is transient as VE-cadherin can recycle on the cell membrane. 128 Regarding the tight junctions, the concentration of tight junctions is enhanced in the arterioles, lower in capillaries and even scarcer in venules, and was associated with enhanced permeability observed in venules. Claudin-5 is the main constituent of tight junctions. Mice lacking claudin-5 displayed enhanced BBB permeability. 129
Besides adherens and tight junctions, focal adhesion cells (FACs) placed close to endothelial cells were shown to play a role in vascular permeability. Lack of adhesion to extracellular matrix impairs endothelial function. Furthermore, growth factors, thrombin, histamine, and bradykinin also disrupt FAC and endothelial cell homeostasis. 130 Thus, endogenous- or exogenous-mediated changes in the endothelium lining structure or the levels of some inflammatory mediators can lead to enhanced permeability, disturbing tissue and organ function.
Changes in blood vessel permeability due to coronaviruses and potential therapeutical targets
Mortality driven by coronaviruses primarily results from lung fluid accumulation, leading to acute respiratory distress syndrome (ARDS).33,131,132 Unbalance between fluid coming in and out of alveoli after blood vessel leakage significantly contributes to lung edema. Accumulated fluid impairs normal air exchanges, that is, tissue oxygenation and CO2 elimination. 133 Coronaviruses change blood vessel permeability in the following way.
SARS-CoV
Both direct and indirect mechanisms mediate the SARS-CoV-induced vascular permeability change. The direct mechanism is thought to be due to a highly hydrophobic protein in their structure, generally termed viroporin, which oligomerize and form hydrophilic pores in the cell membrane lipid bilayer. It was demonstrated that the E protein in SARS-CoV behaves this way in mammalian cells.134,135 Thus, it is possible that the SARS-CoV’s E protein directly interacts with endothelium, enhancing blood vessel permeability. As an indirect mechanism, the SARS-CoV virus infects the respiratory epithelium and several other cells in the body, including enterocytes. 21 As a result, increased intestinal permeability to lipopolysaccharide (LPS) and intestinal bacterial transmigration can enhance permeability in other organs, like the lungs. Drugs that act on these targets may alleviate the respiratory symptoms observed in some SARS-CoV-infected patients.
SARS-CoV-2
Similar to the SARS-CoV virus, the SARS-CoV-2 virus increases blood vessel permeability through indirect and direct mechanisms. As mentioned before, after invading both the epithelial cell type II and endothelial cells in the lungs primarily by binding to the ACE2 receptor and a couple of other proteins placed on the extracellular membrane, the SARS-CoV-2 virus leads to the release of pro-inflammatory cytokines, such as 1L-1β, TNF-α, IL-6, and IFN-γ, and growth factors, like VEGF, from these cells. As a result, immune cell recruitment, enhanced permeability, pyroptosis, epithelial and endothelial cell death, and a further increase in vascular leakage are observed. It has been proposed that these responses are an indirect mechanism of the SARS-CoV-2-induced increase of blood vessel permeability.91,136–139 Corroborating these studies, Xiong et al. 140 demonstrated reduced permeability in the lungs of a murine model of COVID-19 infection by inhibiting IL-1R.
VEGF is also primarily responsible for a vascular phenomenon observed in different organs – for example, lungs, liver, kidneys, and heart – of COVID-19 patients: intussusceptive angiogenesis.141,142 This type of angiogenesis is characterized by blood vessels splitting into two lumens after forming an intraluminal septum (inside to outside growing) mainly due to microthrombi presence, and it was reported to be macrophage-dependent in COVID-19 patients.143,144 In fact, enhanced circulating levels of VEGF were observed in COVID-19 patients and even correlated with disease severity. 145 Intussusceptive angiogenesis may contribute to COVID-19 abnormalities as it disturbs the regular blood flow, leading to further microthrombi formation. 146 Thus, avoiding microthrombi formation and targeting VEGF are potential mechanisms for reducing blood vessel abnormalities, including edema and intussusceptive angiogenesis, in SARS-CoV-2-infected patients. Furthermore, the role of NETs in the enhanced vascular permeability observed in COVID-19 patients has not been determined yet, but as it contributes to the microthrombi formation, 92 targeting NETs is another potential mechanism of reducing blood vessel leakage.
As other potential indirect mechanisms of enhanced blood vessel permeability in the lung tissue, ACE2 receptor endocytosis after SARS-CoV-2 binding may result in increased bradykinin levels, which disrupts vascular permeability 147 and ROS production after toll-like receptor (TLR) activation, with viral RNA binding leading to endothelial cells injury and subsequent blood vessel leakage. 148
As direct mechanisms, Kovacs-Kasa et al. 149 recently suggested that an unidentified heat-labile plasma factor different from thrombin, cytokines, and the complement factors C3a and C5a is responsible for the enhanced vascular permeability observed in SARS-CoV-2-infected patients. In this sense, Robles et al. 150 recently demonstrated that the S spike protein of SARS-CoV-2 can directly enhance permeability in vitro and in vivo by interacting with the α5β1 integrin on endothelial cells. The S spike protein was shown to disturb junctional proteins on endothelial cells, including VE-cadherin and JAM-A, the latter being a tight junctional protein. 151 Corroborating this finding, Lu et al. 152 using a vasculature-on-a-chip platform showed SARS-CoV-2 disrupts VE-cadherin junctions. A similar result was demonstrated by Nader et al. 153 using human endothelial cells in culture, in which they reported downregulated VE-cadherin expression after the S spike protein of SARS-CoV-2 binds to the αvβ3 integrin on endothelial cells. Furthermore, YANG et al. 154 reported a role for the M protein of SARS-CoV-2, enhancing vascular permeability by inducing apoptosis of lung epithelial cells after indirectly interacting with mitochondria. A role for the N protein of SARS-CoV-2 enhancing permeability was demonstrated in mice. 155 Other proteins of SARS-CoV-2 – for example, nsp2, nsp5_c145a, and nsp7 – increased permeability in vitro in endothelial cell cultures. 156 Some of these mechanisms are presented in Figure 3.

Mechanisms of coronaviruses-induced enhanced permeability. In (a), coronavirus proteins (S, E, N, M, and nsps) directly enhance permeability. Some possible mechanisms include (1) E protein acting as a viroporin forming pores in the cell membrane; (2) S protein activates integrins and reduces junctional proteins expression; and (3) M protein acts on mitochondria and leads to cell apoptosis. In (b), indirect mechanisms of coronaviruses-induced enhanced permeability include (1) ACE2R internalization, leading to enhanced circulating bradykinin levels; (2) release of VEGF and cytokines can directly enhance permeability and lead to cell pyroptosis, further enhancing permeability; (3) by the release of an unidentified heat-labile plasma factor; (4) viral RNA binding to the toll-like receptor, leading to ROS production and cell injury. Epithelial and endothelial cell death may increase vascular leakage.
Table 2 contains potential targets of SARS-CoV-2-induced enhanced blood vessel permeability.
Potential targets of drugs to reduce coronaviruses-induced blood vessel permeability dysregulation.
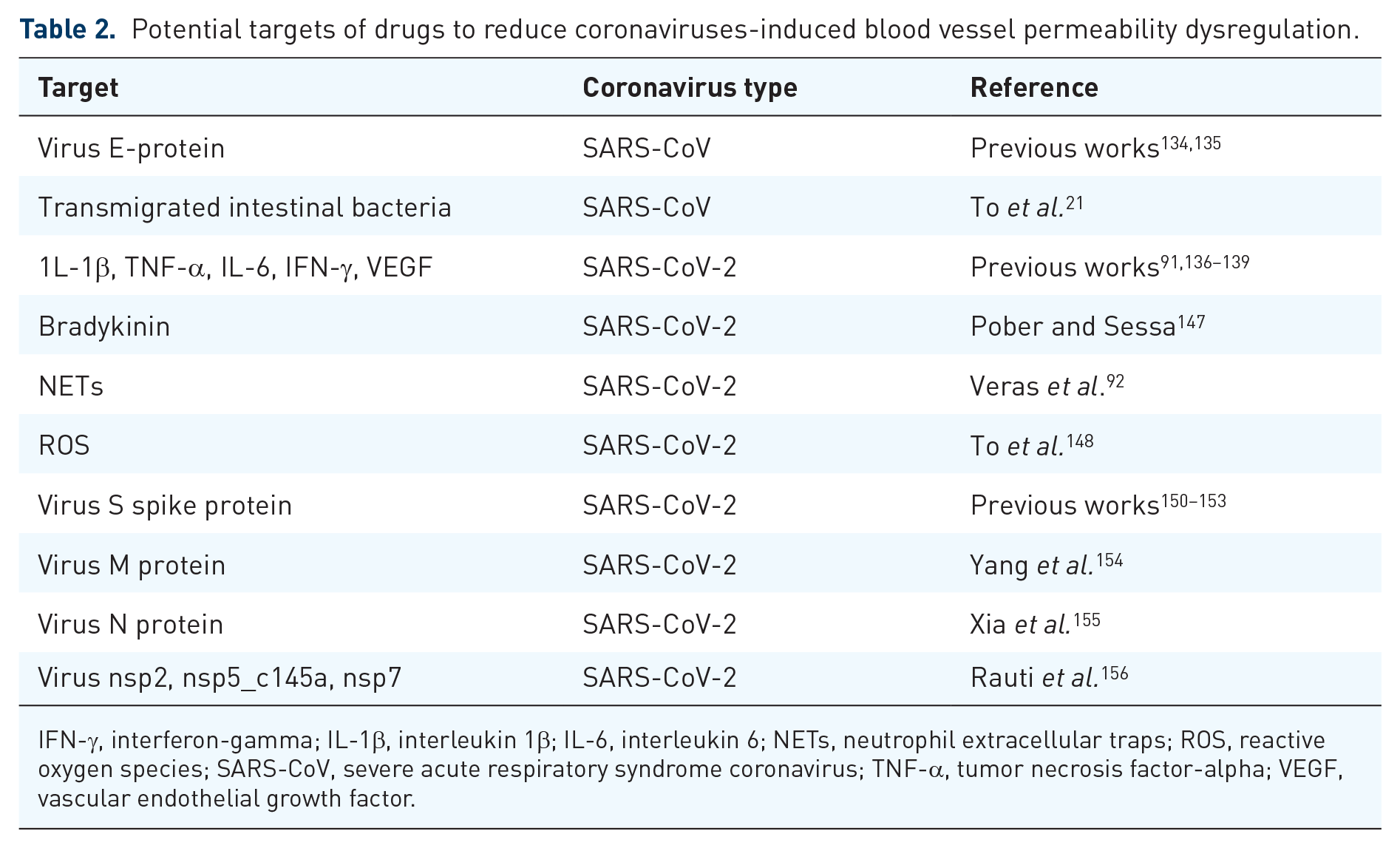
IFN-γ, interferon-gamma; IL-1β, interleukin 1β; IL-6, interleukin 6; NETs, neutrophil extracellular traps; ROS, reactive oxygen species; SARS-CoV, severe acute respiratory syndrome coronavirus; TNF-α, tumor necrosis factor-alpha; VEGF, vascular endothelial growth factor.
Thus, a direct action of the SARS-CoV-2 virus enhancing vascular permeability and an endogenously mediated indirect activity appear to be notable contributors to lung edema observed in patients with COVID-19 and may work as targets for drug action, reducing morbidity and mortality.
Owing to the enhanced blood vessel permeability observed in COVID-19 patients, reduced microvascular blood flow is observed. Koutsiaris et al. 157 reported 39%, 49%, and 47% reductions of axial blood velocity in capillaries and different-sized postcapillary venules, respectively, in the eye microcirculation of COVID-19 patients compared with a non-infected group. Moreover, microvascular dilation was reported in peribronchial and perivascular microvessels (vasa vasorum) of patients who succumbed to severe COVID-19 compared with non-COVID-19 patients. 158 Thus, along with the compromised respiratory function of SARS-CoV-2-infected patients, further tissue ischemia seems to corroborate dysfunction in several organs.
MERS-CoV
The mechanism by which the MERS-CoV virus enhances permeability remains unknown.
Drugs used to reduce the coronaviruses-induced enhanced permeability in the lung
As coronaviruses in larger or lower extensions affect blood vessels through an excessive inflammatory response giving origin to symptoms such as shortness of breathing as a consequence of a disseminated thrombosis and enhanced vascular permeability, a search for treatments that can preserve the blood vessels’ integrity may be vital to preventing the more severe cases of coronavirus-induced diseases. Drugs used in the treatment of coronavirus infections that can modulate the enhanced blood vessel permeability include:
SARS-CoV: there is currently no cure for SARS, and the search for an efficacious vaccine is ongoing. Treatment is primarily supportive and includes steroids to treat lung edema, but large clinical trials to confirm the effectiveness of steroids to treat SARS are still lacking. 159
MERS-CoV: there is no specific treatment for MERS-CoV-induced infection; treatment is given to reduce symptoms. Furthermore, no vaccine prevents or reduces symptoms or mortality due to MERS-CoV infection. 160 Unlike SARS-CoV-infected patients, a multicenter retrospective study showed that MERS-CoV-infected patients who received steroids were more likely to be on a ventilator than those who did not receive it. Besides, patients who got steroids had delayed virus clearance. 161 Thus, unless indicated for other paralleled clinical conditions, steroids must be avoided in patients with MERS-CoV infection.
SARS-CoV-2: among the drugs that target the virus, Food and Drug Administration (FDA)-approved preferred therapy for COVID-19 may include one antibody, sotrovimab, and some antivirals, nirmatrelvir with ritonavir, and remdesivir. As strategies to reduce the effects of the cytokine storm, an anticoagulant, heparin, unless contraindicated, an anti-inflammatory, dexamethasone, and drugs or antibodies that target some components of the cytokine storm, particularly baricitinib, tocilizumab, or sarilumab. Treatment changes according to the disease severity. 162
Clinically approved drugs or those in clinical trials intended to reduce the SARS-CoV-2-induced vascular permeability increase in lungs include glucocorticoids, anakinra, and tocilizumab. Glucocorticoids are a class of hormones that have immunosuppressive and anti-inflammatory properties, inhibiting pro-inflammatory genes in immune cells through the glucocorticoid receptor (GR), which is present in practically every cell in the human body. This gene inhibition causes the downregulation of cytokines directly and indirectly related to the enhanced vascular permeability such as IL-1β and IL-1α, MCP-1, and macrophage inflammatory protein-2 (MIP-2). Also, it induces apoptosis of polymorphonuclear leukocytes (PMNs), like neutrophils, basophils, eosinophils, and T-cells, resulting in the subsequent reduction of the inflammatory state. 163 PMNs cause a barrier dysfunction in adherent junctions through proteolysis of VE-cadherin in endothelial cells, which in turn causes hyperpermeability of the vascular tissue. 164 By utilizing pharmacological properties that combat this hyperinflammatory state and subsequent vascular hyperpermeability, glucocorticoids are effective in lowering the mortality of COVID-19 patients who require invasive treatment, with a study showing a difference in the death rate by as much as 29.3% in patients who received dexamethasone, a glucocorticoid medication, versus 41.4% of the patients who received the usual invasive care without the medication. 165 Moreover, it has been reported that neutrophil infiltration in the lungs is correlated with worsened prognosis in COVID-19 patients, supporting the use of this class of medication due to its immunosuppressive effects. 166 Despite demonstrated corticosteroid-induced improvements in oxygenation rate in ARDS subjects, 167 oxygenation rate may be an indirect form of measuring lung edema. In this context, a case report demonstrated corticosteroids improved oxygenation in patients with COVID-19; 168 other case reports observed corticosteroids diminished cerebral edema in these patients.169,170 It is worth mentioning that corticosteroids may present different outcomes in ARDS subjects depending on several factors, including the timing of initiation of treatment, the origin of ARDS, type and dose of corticosteroid, and among others (for a review on corticosteroids and ARDS, see Hussain et al. 171 ).
Continuing the approach to target the acute leaky response induced by cytokines, current studies are being conducted regarding IL-1 receptor antagonists, such as anakinra, and their effectiveness in treating COVID-19 symptoms by blocking the inflammatory cascade caused by the cytokines IL-1β and IL-1α. Among these studies, Xiong et al. 140 used a murine model of COVID-19 infection and reported reduced permeability in the lungs of animals treated with anakinra. Corroborating this result, but in a clinical setting, Cavalli et al. 172 retrospectively compared patients administered a high dose of anakinra and standard treatment with a control group that was given only standard treatment. The population included consecutive patients with COVID-19, moderate-to-severe ARDS, or hyperinflammation (defined as serum C-reactive protein ⩾100 mg/l, ferritin ⩾900 ng/ml, or both). As a result, at 21 days, survival was 90% in the high-dose anakinra group and 56% in the standard treatment group, and mechanical ventilation-free survival was 72% in the anakinra group versus 50% in the standard treatment. Although animating, a drawback of this study was that it contained few individuals in each group (29 in the anakinra group and 16 in the control group). Owing to how IL-1β has been shown to affect vascular integrity through reduced VE-cadherin expression in endothelial cells, especially in the lungs, which can turn into pneumonitis by the inflammatory edema, pursuing more extensive clinical trials regarding IL-1 receptor antagonists is highly recommended. 173 Another robust study showed the efficacy of IL-1 receptor antagonists in patients who received anakinra, as evidenced by a lower need for invasive mechanical ventilation, decreased need for oxygen supply in patients who did not need invasive mechanical ventilation, and lower levels of reactive C-protein, an indicator of hyperinflammation, compared with a group not receiving the drug. 174
Another class of medication currently under clinical trials and is showing promising results is the class of IL-6 antagonists. These drugs specifically target elevated vessel permeability, among other inflammatory issues caused by the disease, possibly attenuating pulmonary edema. IL-6 is a reliable marker of patient outcome and disease severity and correlates with cytokine release syndrome (CRS) and lung complications in COVID-19 patients. 175 With this said, one agent that functions as an IL-6 antagonist, tocilizumab, has been associated with a lower mortality rate in COVID-19 patients requiring mechanical ventilation and faster hospital discharge. 176
Drugs discussed in this section and related clinical assays performed on patients with COVID-19 are presented in Table 3.
Clinically assayed drugs that reduce vascular leakage in the lungs to treat patients with COVID-19.

Conclusion
Enhanced blood vessel permeability greatly contributes to symptoms and mortality caused by coronaviruses. Prostaglandins, leukotrienes, and thromboxanes, IL-1 and IL-6, are known to dysregulate the endothelial–alveolar barrier function, resulting in increased permeability. Thus, they are potential targets to reduce the enhanced coronavirus-induced permeability. Drugs that attenuate the activity of phospholipase A2, IL-1, and IL-6 are notable tools for alleviating symptoms and mortality in patients infected with coronaviruses. Thus, the search for new drugs or the repositioning of others that interfere with these targets promises to be good strategies for reducing coronavirus-induced morbidity and mortality.
Literature search
The electronic platforms used to search for articles used to write this review were PubMed and Google scholar. Keywords related to each topic were applied alone or combined the following way: first topic, ‘Coronaviruses and diseases’: SARS, MERS, SARS-CoV-2, Middle East respiratory coronavirus, severe acute respiratory syndrome coronavirus, and COVID-19; topic ‘blood vessel permeability’: blood vessel permeability, blood vessel leakage, blood vessel and edema, and edema and endothelial junctions; topics ‘Changes in blood vessel permeability due to coronaviruses and potential therapeutical targets’ and ‘Drugs used to reduce the coronaviruses-induced enhanced permeability in lung’: blood vessels and coronavirus, blood vessels and SARS, blood vessels and severe acute respiratory syndrome coronavirus, SARS and edema, severe acute respiratory syndrome coronavirus and edema, blood vessels and MERS, blood vessels and Middle East respiratory coronavirus, MERS and edema, Middle East respiratory coronavirus and edema, blood vessels and SARS-CoV-2, blood vessels and COVID-19, SARS-CoV-2 and edema, and COVID-19 and edema. Both original and review articles were considered. Clinical trials were searched for on the following websites: https://clinicaltrials.gov/ and https://www.clinicaltrialsregister.eu/.