
Abstract
Background:
In chronic obstructive pulmonary disease (COPD), both the time needed for patients to gain symptom improvement with long-acting bronchodilator therapy and whether an early response is predictive of a sustained response is unknown. This study aimed to investigate how quickly meaningful symptom responses are seen in patients with COPD with bronchodilator therapy and whether these responses are sustained.
Methods:
Early MAXimisation of bronchodilation for improving COPD stability (EMAX) was a 24-week, double-blind, double-dummy, parallel-group trial that randomised patients to umeclidinium/vilanterol (UMEC/VI), umeclidinium or salmeterol. Daily Evaluating Respiratory Symptoms in COPD (E-RS:COPD) score and rescue salbutamol use were captured via an electronic diary and analysed initially in 4-weekly periods. Post hoc analyses assessed change from baseline in daily E-RS:COPD score and rescue medication use weekly (Weeks 1–8), and association between E-RS:COPD responder status at Weeks 1–4 and later time points.
Results:
In the intent-to-treat population (n = 2425), reductions from baseline in E-RS:COPD scores and rescue medication use were apparent from Day 2 with all treatments. Treatment differences for UMEC/VI versus either monotherapy plateaued by Week 4–8 and were sustained at Weeks 21–24; improvements were consistently greater with UMEC/VI. For all treatments, most patients (60–85%) retained their Weeks 1–4 E-RS:COPD responder/non-responder status at Weeks 21−24. Among patients receiving UMEC/VI who were E-RS:COPD responders at Weeks 1–4, 70% were responders at Weeks 21–24.
Conclusion:
Patients with symptomatic COPD had greater potential for early symptom improvements with UMEC/VI versus either monotherapy. This benefit was generally maintained for 24 weeks. Early monitoring of treatment response can provide clinicians with an early indication of a patient’s likely longer-term response to prescribed bronchodilator treatment and will facilitate appropriate early adjustments in care.
Clinical Trial Registration:
NCT03034915, 2016-002513-22 (EudraCT Number).
The reviews of this paper are available via the supplemental material section.
Introduction
The recommended first-line treatment for patients with symptomatic chronic obstructive pulmonary disease (COPD) at low risk of exacerbations is monotherapy with a long-acting muscarinic antagonist (LAMA) or a long-acting beta-agonist (LABA) bronchodilator. Due to their differing mechanisms of action, combining a LAMA (which mediates bronchodilation through antagonism of muscarinic acetylcholine M3 receptors) with a LABA (which mediates bronchodilation through activation of β2-adrenoceptors) maximises bronchodilation with greater efficacy than either monotherapy alone. 1 Therefore, LAMA/LABA dual therapy is considered appropriate in patients who experience severe breathlessness or have a high risk of exacerbations. 2 Managing respiratory symptoms is a key treatment goal in COPD 2 ; however, the time taken for patients with COPD to achieve a maximal symptomatic response to bronchodilator therapy is not well understood. Furthermore, it is not known if an early (e.g. within the first month), clinically meaningful response to bronchodilator therapy is predictive of a sustained response and beneficial future long-term outcomes.
It is important that validated, reliable tools are used to monitor the effect of treatment on symptoms. The Evaluating Respiratory Symptoms in COPD (E-RS:COPD) is a patient-reported symptom scale that quantifies the symptoms of COPD to give a total score (range 0–40) as a summation of three subdomain scores [breathlessness (0–17), cough and sputum (0–11) and chest symptoms (0–12)]. It can be used to monitor changes over time, with a decrease in score indicating an improvement in symptoms. 3 The E-RS:COPD has shown evidence of content validity, reliability and responsiveness in patients with COPD,3,4 and the US Food and Drug Administration and the European Medicines Agency support its use as an endpoint in clinical trials of COPD.5,6 Unlike other patient-reported measures commonly used in COPD, such as the St George’s Respiratory Questionnaire 7 and the Transition Dyspnoea Index, 8 the E-RS:COPD is measured daily as a diary, and is therefore well suited to document the time to response following the initiation of therapy.
The Early MAXimisation of bronchodilation for improving COPD stability (EMAX) trial assessed the LAMA/LABA umeclidinium/vilanterol (UMEC/VI) versus UMEC or salmeterol (SAL) in symptomatic, low exacerbation risk patients with COPD who were not receiving inhaled corticosteroids (ICS). The trial demonstrated greater improvements in lung function and symptoms at 6 months with UMEC/VI versus UMEC or SAL monotherapy. 9 This analysis investigated the time to symptom improvement and consistency of improvement in daily symptoms according to two measures, the E-RS:COPD score and rescue medication use.
Methods
Study design, patients and outcomes
The 24-week, multicentre, randomised, double-blind, double-dummy, 3-arm parallel-group EMAX trial (NCT03034915; GSK study 201749), conducted between June 2017 and June 2018, randomised patients 1:1:1 to once-daily fixed-dose combination UMEC/VI 62.5/25 µg via the ELLIPTA inhaler and twice-daily placebo via the DISKUS inhaler, once-daily UMEC (62.5 µg) via ELLIPTA and twice daily placebo via DISKUS or twice-daily SAL (50 µg) via DISKUS and once-daily placebo via ELLIPTA. Patients were treated for 24 weeks following a 4-week run-in period. The study was performed according to the Declaration of Helsinki and received appropriate ethical approval. All patients provided written informed consent. This manuscript conforms to the CONSORT guidelines for publication of randomised controlled trials (Supplemental Table 1).
Full details of the study design and patient population have been reported previously. 9 In brief, eligible patients were ⩾40 years of age, current/former smokers (⩾10 pack-years smoking history), with a COPD diagnosis (American Thoracic Society/European Respiratory Society definition), pre- and post-salbutamol forced expiratory volume in 1 second (FEV1)/forced vital capacity ratio of < 0.7, post-salbutamol FEV1 of ⩾30–⩽80% predicted [Global Initiative for Chronic Obstructive Lung Disease (GOLD) stage 2/3], COPD Assessment Test (CAT) score of ⩾10, with ⩽1 moderate exacerbation and no severe exacerbations in the previous year. Before screening and during a 4-week run-in period, bronchodilator maintenance therapy was limited to none or one long-acting bronchodilator (a LAMA or a LABA).
All patients were required to be free of ICS and ICS/LABA for ⩾6 weeks and free of LAMA/LABA for ⩾2 weeks prior to run-in. As-needed salbutamol was allowed throughout all study phases.
Daily symptoms were evaluated using the E-RS:COPD and rescue salbutamol use, which were both captured via an electronic diary. In pre-specified analyses, least squares (LS) mean change from baseline in daily E-RS:COPD total score and subdomain scores, and rescue medication use, were analysed in 4-weekly periods. E-RS:COPD response, defined as a ⩾2-point decrease in E-RS:COPD total score from baseline, 3 was analysed in 4-weekly periods. Post hoc analyses included LS mean change from baseline in daily E-RS:COPD total score and rescue medication use (puffs/day) analysed weekly from Weeks 1 to 8, and responder analyses to determine whether an E-RS:COPD response/non-response at Weeks 1–4 was associated with a sustained response/non-response at each subsequent 4-week period.
Statistical analyses
Results are presented for the intent-to-treat (ITT) population, consisting of all randomised patients who received ⩾1 dose of study treatment. Daily E-RS:COPD score and rescue medication use analyses were descriptive only. Weekly and 4-weekly E-RS:COPD scores and rescue medication use were analysed using mixed model repeated measures, with co-variates of baseline E-RS:COPD score or rescue medication use and geographical region, number of bronchodilators (0 or 1) per day during run-in, weekly or 4-weekly period, treatment, weekly or 4-weekly period by baseline and weekly or 4-weekly period by treatment interaction. Responder analyses with corresponding odds ratios (OR) and 95% confidence intervals (CIs) were performed using a generalised linear mixed model with treatment as an explanatory variable and 4-weekly period, baseline E-RS:COPD score, number of bronchodilators per day during run-in, geographical region, 4-weekly period by baseline and 4-weekly period by treatment interactions included as covariates.
Results
Patient demographics and baseline characteristics
In the ITT population (n = 2425), patient demographics and baseline characteristics were similar between treatments (Table 1). The majority of patients were using one LAMA or LABA during the run-in period. At baseline, the mean [standard deviation (SD)] E-RS:COPD total score was 10.6 (5.7) and E-RS:COPD subdomain scores were 5.2 (3.2), 3.0 (1.6) and 2.3 (1.7) for the breathlessness, cough and sputum, and chest symptom subdomains, respectively. Mean (SD) baseline salbutamol rescue use was 2.2 (2.5) puffs/day.
Patient demographics and baseline characteristics.

Number of exacerbations requiring oral or systemic corticosteroids and/or antibiotics (moderate) in 12 months prior to screening [patients with > 1 moderate exacerbation or with a severe exacerbation (requiring hospitalisation) were excluded].
An additional 4 (<1%) patients with GOLD grade 1 were randomised (UMEC n = 3; SAL n = 1).
Higher scores indicate more severe symptoms.
Percentage of rescue-free days from Day -28 to Day -1 inclusive.
CAT, COPD Assessment Test; COPD, chronic obstructive pulmonary disease; E-RS:COPD, Evaluating Respiratory Symptoms in COPD; FEV1, forced expiratory volume in 1 second; GOLD, Global Initiative for Chronic Obstructive Lung Disease; SAL, salmeterol; SD, standard deviation; UMEC, umeclidinium; VI, vilanterol.
Changes from baseline in E-RS:COPD score
Reductions from baseline in E-RS:COPD total score were apparent from Day 2 with all treatments and the largest improvements were observed with UMEC/VI, followed by UMEC and SAL (Figure 1a). Similar results were seen with UMEC/VI versus either monotherapy for E-RS:COPD breathlessness and chest symptoms subdomain scores, whereas for cough and sputum scores, gradual improvements were seen through 24 weeks (Supplemental Figure 1). Reductions from baseline with UMEC/VI versus monotherapy were statistically significant from Week 1 (versus UMEC: 0.45, p = 0.001; versus SAL 0.52, p < 0.001) and remained statistically significantly greater for UMEC/VI versus both UMEC and SAL at Weeks 4 and 8 (Figure 1b). Treatment differences (UMEC/VI versus either monotherapy) plateaued by Weeks 4 to 8 and were sustained over 24 weeks. The mean improvement in E-RS:COPD total score did not reach the minimal clinically important difference (MCID) of 2.0, 3 with LS mean improvements from baseline at Week 4 of 1.15, 0.72 and 0.38 and at Week 8 of 1.41, 0.96 and 0.66 for UMEC/VI, UMEC and SAL respectively. For the E-RS:COPD individual subdomain scores, significant reductions in E-RS:COPD breathlessness and chest symptoms scores were apparent from Week 1 in patients receiving UMEC/VI compared with UMEC and SAL, and in E-RS:COPD cough and sputum scores at Weeks 4 and 8 for UMEC/VI versus SAL (Supplemental Figure 2). At Weeks 21–24 there was a 1.52 reduction from baseline in E-RS:COPD total score with UMEC/VI; for the E-RS:COPD subdomain scores, there were 0.67, 0.45 and 0.39 reductions in breathlessness, cough and sputum, and chest symptoms scores, respectively (Figures 1b and Supplemental Figure 2b).

Daily mean change from baselinea over time in E-RS:COPD total score (a) and LS mean change from baseline in E-RS:COPD total score (b).
E-RS:COPD total score responder analyses
At Weeks 1–4 the highest proportion of E-RS:COPD total score responders were seen in the UMEC/VI treatment arm compared with UMEC and SAL, with the odds of a response significantly favouring UMEC/VI [OR (95% CI): UMEC/VI versus UMEC 1.26 (1.00, 1.58), p = 0.047; UMEC/VI versus SAL 1.37 (1.09, 1.73), p = 0.006] (Figure 2). The baseline characteristics of patients who were E-RS:COPD total score responders and non-responders at Weeks 1–4 were similar except that a greater proportion of E-RS:COPD responders were female, current smokers, maintenance-naïve, had CAT scores ⩾20 at screening and were more symptomatic with higher E-RS:COPD scores at baseline (Supplemental Table 2). At Weeks 21–24, the proportion of E-RS:COPD total score responders increased in all treatment groups with the odds of a response significantly favouring UMEC/VI [OR (95% CI): UMEC/VI versus UMEC 1.52 (1.22, 1.89); p < 0.001; UMEC/VI versus SAL 1.53 (1.23, 1.90); p < 0.001] (Figure 2).

E-RS:COPD total score responders (⩾2-point reduction from baseline)
For all treatments, most patients who were E-RS:COPD responders or non-responders at Weeks 1−4 maintained their responder or non-responder status at Weeks 21−24 (Tables 2 and 3). With UMEC/VI, a small proportion (22%) of patients who were non-responders at Weeks 1−4 had become responders at Weeks 21−24 (Table 3). E-RS:COPD responders at Weeks 1–4 were significantly more likely than non-responders at Weeks 1–4 to be responders at Weeks 21–24 [OR (95% CI): 7.71 (6.24, 9.52); p < 0.001]. Over half (57%, n = 134/236) of E-RS:COPD responders at Weeks 1–4 receiving UMEC/VI maintained their response through each subsequent 4-week period, compared with 46% (n = 92/202) and 44% (n = 82/188) of responders receiving UMEC or SAL, respectively (Table 2). For all treatments, most E-RS:COPD non-responders at Weeks 1–4 remained non-responsive at subsequent time points (Table 3).
Proportion of E-RS:COPD responders a at Weeks 1–4 who sustained their response.
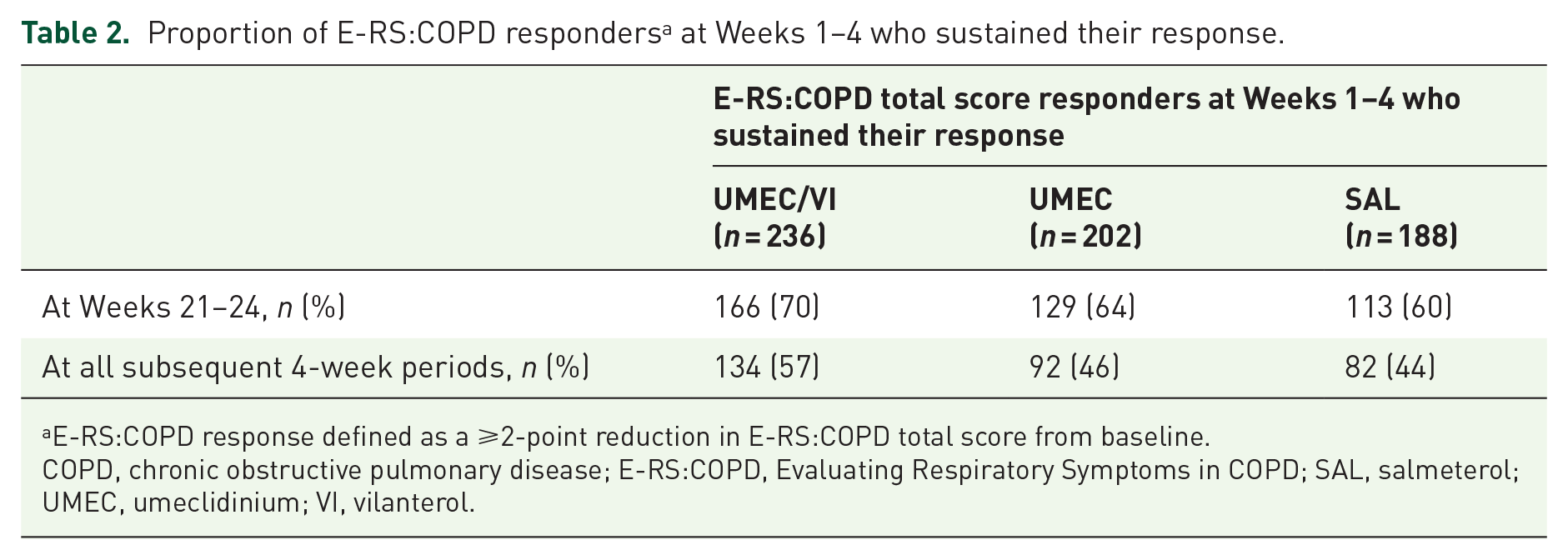
E-RS:COPD response defined as a ⩾2-point reduction in E-RS:COPD total score from baseline.
COPD, chronic obstructive pulmonary disease; E-RS:COPD, Evaluating Respiratory Symptoms in COPD; SAL, salmeterol; UMEC, umeclidinium; VI, vilanterol.
Proportion of E-RS:COPD non-responders a at Weeks 1–4 who sustained their response.
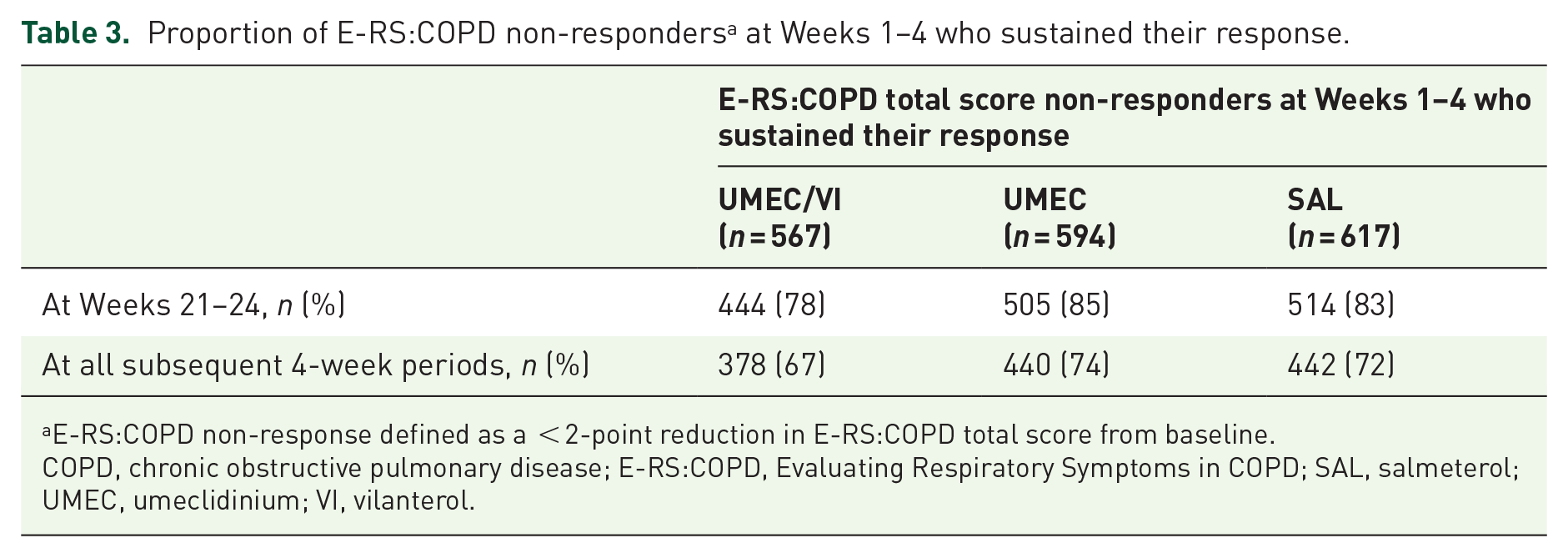
E-RS:COPD non-response defined as a < 2-point reduction in E-RS:COPD total score from baseline.
COPD, chronic obstructive pulmonary disease; E-RS:COPD, Evaluating Respiratory Symptoms in COPD; SAL, salmeterol; UMEC, umeclidinium; VI, vilanterol.
Changes in rescue medication use
Reductions from baseline in rescue medication use were apparent from Day 2 with all treatments, established by Weeks 4–8 and sustained over 24 weeks (Figure 3a). The greatest reductions in rescue medication use were seen with UMEC/VI and the reduction with SAL was numerically greater than that with UMEC (Figure 3). Reductions from baseline in rescue use with UMEC/VI versus monotherapy were statistically significant from Week 1 (versus both UMEC and SAL: 0.20 puff/day, p = 0.002) and remained significantly greater for UMEC/VI versus both UMEC and SAL at Weeks 4 and 8. At Weeks 21–24 there was a 27% (0.6 puffs/day) reduction from baseline in rescue medication use with UMEC/VI (Figure 3b).
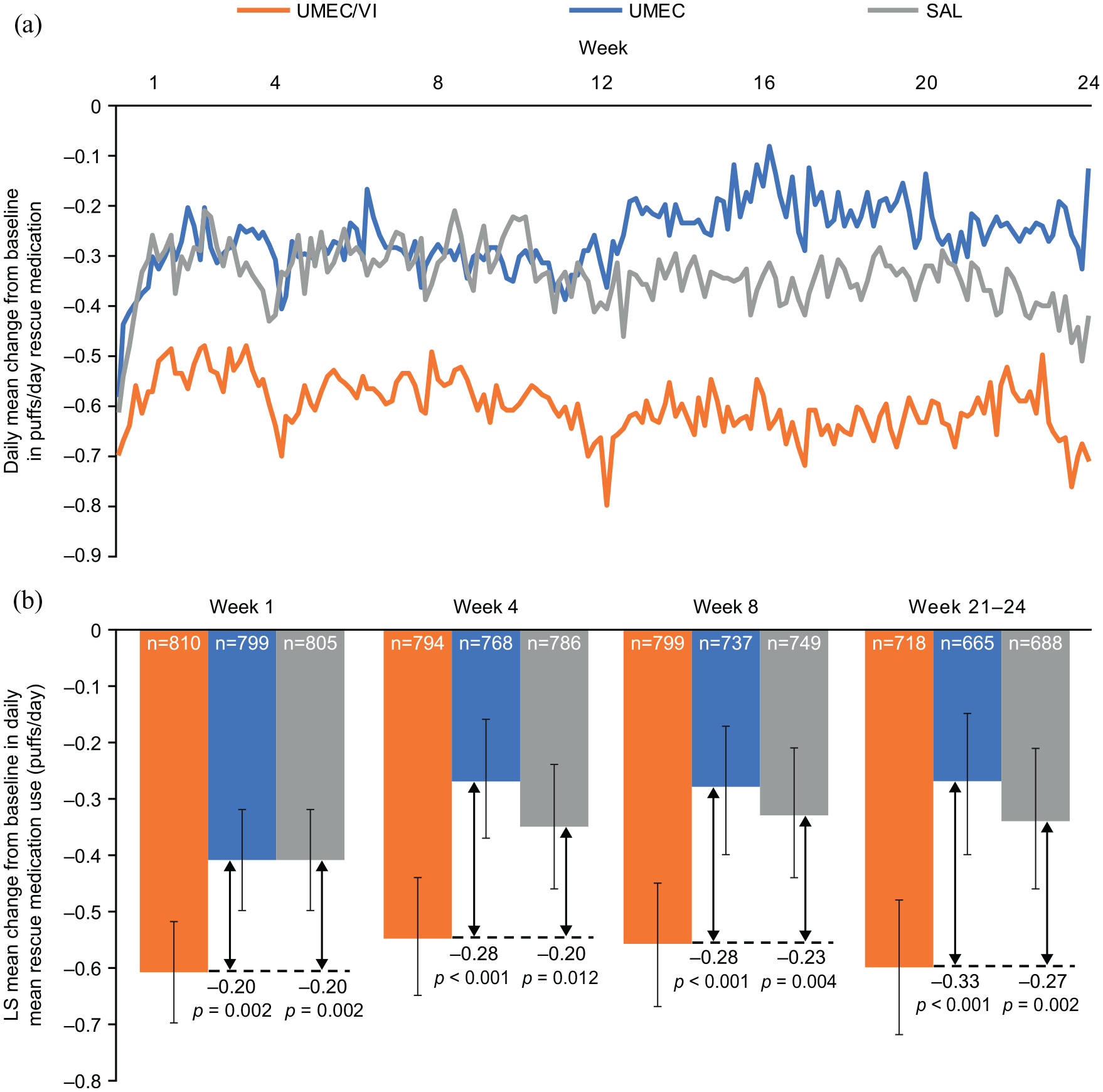
Daily mean change from baseline rescue medication use (puffs/day) (a) LS mean change from baseline rescue medication use (puffs/day) (b).
At Weeks 1–4, the LS mean (95% CI) change from baseline in percentage of rescue-free days was 10.8% (8.8, 12.8), 5.4% (3.3, 7.4) and 6.7% (4.5, 8.7) for UMEC/VI, UMEC and SAL, respectively. The improvements were sustained throughout the study and at Weeks 21–24 the LS mean (95% CI) change from baseline in percentage of rescue-free days was 12.4% (9.9, 14.9), 7.0% (4.4, 9.5) and 8.9% (6.4, 11.4) for UMEC/VI, UMEC and SAL, respectively. At all 4-week intervals the improvement from baseline was significantly greater for UMEC/VI versus UMEC and versus SAL [between treatment differences (95% CI) at Weeks 21–24: UMEC/VI versus UMEC, 5.4% (1.9, 9.0), p = 0.003; UMEC/VI versus SAL, 3.5% (0.0, 7.0), p = 0.049].
Discussion
In this analysis of the EMAX trial, improvements in symptoms and rescue medication use were observed in all treatment groups within the first few days of treatment initiation. Treatment differences for UMEC/VI versus either monotherapy plateaued by Week 4 to 8 and were sustained at Weeks 21–24; improvements were consistently greater with UMEC/VI. Consistent with previous reports of the benefits of dual bronchodilation compared with mono-bronchodilator therapy,10–13 the improvements in E-RS:COPD score and rescue medication use were consistently greater with UMEC/VI compared with either monotherapy in all study periods. This early improvement in symptom response was more likely to be sustained over time with UMEC/VI compared with either UMEC or SAL.
Early treatment response at Weeks 1–4 as measured by E-RS:COPD total score was associated with a maintained response over subsequent weeks. For all treatments, more E-RS:COPD total score responders were observed at Weeks 21–24 than at Weeks 1–4; however, overall a non-response within 1 month of treatment was associated with continuing to be a non-responder at subsequent time points. This suggests that long-term trials may not provide additional information compared with shorter trials when determining differences in symptomatic response between bronchodilator therapies, although longer-term trials will still be needed to assess disease modification, exacerbation outcomes and safety. These results also reinforce that in clinical practice, physicians should evaluate symptom improvement after approximately 4–8 weeks of treatment to gain an early indication of whether a patient is likely to respond to their prescribed bronchodilator treatment and identify patients who require further treatment modifications.
Approximately two-thirds of patients in the EMAX trial used daily maintenance LAMA or LABA during the run-in period and, as such, the early changes in E-RS:COPD reflect improvements over and above that achieved on baseline bronchodilators and short-acting beta-agonist (SABA). Although the change from baseline in E-RS:COPD total score did not meet the established MCID of 2.0, this method of reporting outcomes does not provide an indication of the proportion of patients who improved by more than the MCID. 14 However, the ORs were 1.52 and 1.53 for the E-RS:COPD total score responder analysis of UMEC/VI versus UMEC and SAL, respectively, demonstrating that there was approximately a 50% greater chance in the odds for patients having clinically significant improvement with UMEC/VI than with UMEC or SAL.
Monitoring of rescue medication use is common in patients with asthma in clinical trials and in clinical practice.15,16 It has also been identified as a marker of symptomatic benefit in COPD trials, 17 with rescue-free days and puffs/day being important indicators of COPD clinical stability over time. 18
The EMAX population were relatively low users of rescue SABA at baseline (2.2 puffs/daily) compared with previous studies that have reported a mean baseline use of approximately 4 puffs/day among typical clinical trial patients with symptomatic COPD. 19 Patients receiving UMEC/VI achieved a 0.6 puffs/day reduction from baseline in SABA use by the end of the study (equating to 27%), with this reduction apparent within the first few days of treatment. A systematic review of multiple clinical trials also demonstrated clinically relevant changes in health status and exacerbation risk with similar changes in rescue use from baseline (0.6–1.0 puffs/day). 20 Patients on UMEC/VI also had a 12% increase in rescue-free days by Weeks 21–24, with an 11% improvement seen as early as Weeks 1–4. Although there is no established MCID for rescue medication use in COPD, the improvements at both these time points exceeded the ⩾8.3% difference identified as clinically important in a previous study. 18
Rescue medication use can be quickly and easily assessed in clinical practice and this study and previous evidence suggests that reduction in rescue medication use is associated with improvements in E-RS:COPD total score, 21 which is a validated, established outcome for measuring COPD symptoms. 3 However, another prospective analysis of the EMAX trial suggests that the relationship is complex and warrants further investigation. 22
There are some study limitations that should be considered when interpreting these results. The study compared UMEC/VI and UMEC, which are administered once daily, with SAL, which is administered twice daily (VI could not be used because it is not licensed as a monotherapy). Patients were permitted to use a single LAMA or LABA during the study run-in period, so most patients were switched from their existing inhaled maintenance treatment to study treatment; therefore, the treatment effects may be underestimated compared with the effect that might be seen in patients not receiving maintenance treatment before joining the study. The EMAX study was 24 weeks in duration; longer-term studies are required to determine whether the responses measured at the end of this study are sustained for longer treatment durations. In addition, this was a post hoc analysis and as such the results should be confirmed prospectively.
Conclusions
Dual bronchodilator treatment with once-daily UMEC/VI is associated with a greater early and sustained symptomatic benefit than UMEC or SAL, with the symptom improvements with dual therapy seen from the first few days of treatment initiation. These findings suggest that monitoring patients’ symptom response to bronchodilator treatment may provide valuable prognostic information on longer-term treatment responses and aid earlier treatment decisions.
Supplemental Material
Author_Response – Supplemental material for Early and sustained symptom improvement with umeclidinium/vilanterol versus monotherapy in COPD: a post hoc analysis of the EMAX randomised controlled trial
Supplemental material, Author_Response for Early and sustained symptom improvement with umeclidinium/vilanterol versus monotherapy in COPD: a post hoc analysis of the EMAX randomised controlled trial by Edward M Kerwin, Isabelle H Boucot, Claus F Vogelmeier, Francois Maltais, Ian P Naya, Lee Tombs, Paul W Jones, David A Lipson, Tom Keeley and Leif Bjermer in Therapeutic Advances in Respiratory Disease
Supplemental Material
Reviewer_1_v.1 – Supplemental material for Early and sustained symptom improvement with umeclidinium/vilanterol versus monotherapy in COPD: a post hoc analysis of the EMAX randomised controlled trial
Supplemental material, Reviewer_1_v.1 for Early and sustained symptom improvement with umeclidinium/vilanterol versus monotherapy in COPD: a post hoc analysis of the EMAX randomised controlled trial by Edward M Kerwin, Isabelle H Boucot, Claus F Vogelmeier, Francois Maltais, Ian P Naya, Lee Tombs, Paul W Jones, David A Lipson, Tom Keeley and Leif Bjermer in Therapeutic Advances in Respiratory Disease
Supplemental Material
Reviewer_2_v.1 – Supplemental material for Early and sustained symptom improvement with umeclidinium/vilanterol versus monotherapy in COPD: a post hoc analysis of the EMAX randomised controlled trial
Supplemental material, Reviewer_2_v.1 for Early and sustained symptom improvement with umeclidinium/vilanterol versus monotherapy in COPD: a post hoc analysis of the EMAX randomised controlled trial by Edward M Kerwin, Isabelle H Boucot, Claus F Vogelmeier, Francois Maltais, Ian P Naya, Lee Tombs, Paul W Jones, David A Lipson, Tom Keeley and Leif Bjermer in Therapeutic Advances in Respiratory Disease
Supplemental Material
Supplementary_data – Supplemental material for Early and sustained symptom improvement with umeclidinium/vilanterol versus monotherapy in COPD: a post hoc analysis of the EMAX randomised controlled trial
Supplemental material, Supplementary_data for Early and sustained symptom improvement with umeclidinium/vilanterol versus monotherapy in COPD: a post hoc analysis of the EMAX randomised controlled trial by Edward M Kerwin, Isabelle H Boucot, Claus F Vogelmeier, Francois Maltais, Ian P Naya, Lee Tombs, Paul W Jones, David A Lipson, Tom Keeley and Leif Bjermer in Therapeutic Advances in Respiratory Disease
Supplemental Material
Supp_Figure_1_HR – Supplemental material for Early and sustained symptom improvement with umeclidinium/vilanterol versus monotherapy in COPD: a post hoc analysis of the EMAX randomised controlled trial
Supplemental material, Supp_Figure_1_HR for Early and sustained symptom improvement with umeclidinium/vilanterol versus monotherapy in COPD: a post hoc analysis of the EMAX randomised controlled trial by Edward M Kerwin, Isabelle H Boucot, Claus F Vogelmeier, Francois Maltais, Ian P Naya, Lee Tombs, Paul W Jones, David A Lipson, Tom Keeley and Leif Bjermer in Therapeutic Advances in Respiratory Disease
Supplemental Material
Supp_Figure_2_HR – Supplemental material for Early and sustained symptom improvement with umeclidinium/vilanterol versus monotherapy in COPD: a post hoc analysis of the EMAX randomised controlled trial
Supplemental material, Supp_Figure_2_HR for Early and sustained symptom improvement with umeclidinium/vilanterol versus monotherapy in COPD: a post hoc analysis of the EMAX randomised controlled trial by Edward M Kerwin, Isabelle H Boucot, Claus F Vogelmeier, Francois Maltais, Ian P Naya, Lee Tombs, Paul W Jones, David A Lipson, Tom Keeley and Leif Bjermer in Therapeutic Advances in Respiratory Disease
Footnotes
Acknowledgements
The authors would like to thank Chris Compton (Global Specialty & Primary Care, GSK, Brentford, Middlesex, UK) who was involved in the conception and design of the study, data analysis and interpretation and reviewed the manuscript. Editorial support (in the form of writing assistance, assembling figures, collating author comments, grammatical editing, and referencing) was provided by Katie White, PhD, of Fishawack Indicia Ltd, UK, and was funded by GSK.
Author contribution(s)
Conflict of interest statement
EMK has served on advisory boards, speaker panels or received travel reimbursement for Amphastar, AstraZeneca, Boehringer Ingelheim, GSK, Mylan, Novartis, Pearl, Sunovion, Teva and Theravance, and has received consulting fees from Cipla and GSK. IHB, DAL, TK and PWJ are employees of GSK and hold stocks and shares in GSK. CFV has received grants from AstraZeneca, Boehringer Ingelheim, Chiesi, GSK, Grifols, Mundipharma, Novartis and the German Federal Ministry of Education and Research (BMBF) Competence Network Asthma and COPD (ASCONET), and has received personal fees from AstraZeneca, Boehringer Ingelheim, Berlin Chemie/Menarini, Chiesi, Cipla, CSL Behring, GSK, Grifols, MedUpdate, Mundipharma, Novartis, Nuvaira and Teva. FM has received research grants for participating in multicentre trials for AstraZeneca, Boehringer Ingelheim, GSK, Sanofi and Novartis, and has received unrestricted research grants and personal fees from Boehringer Ingelheim, Grifols and Novartis. IPN was an employee of GSK at the time of the study, holds stocks and shares in GSK and is a contingent worker on assignment at AstraZeneca. LT is a contingent worker on assignment at GSK. LB has received honoraria for giving a lecture or attending an advisory board for Airsonett, ALK-Abelló, AstraZeneca, Boehringer Ingelheim, Chiesi, GSK, Meda, Novartis and Teva.
ELLIPTA and DISKUS are owned by/licensed to the GSK group of companies.
Data availability
Ethics approval and informed consent
The study was performed according to the Declaration of Helsinki and received appropriate ethical approval. All patients enrolled in EMAX provided written informed consent.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by GlaxoSmithKline (GSK study 201749; NCT03034915). The funders of the study had a role in the study design, data analysis, data interpretation, and writing of the report.
Supplemental material
The reviews of this paper are available via the supplemental material section.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.