
Abstract
Patients with advanced epidermal growth factor receptor (EGFR) mutant non-small cell lung cancer (NSCLC) are particularly sensitive to treatment with first- or second-generation EGFR tyrosine kinase inhibitors such as gefitinib, erlotinib and afatinib, which block the cell-signaling pathways that drive the growth of tumor cells. Unfortunately, the majority of patients develop resistance to them after a median duration of response of around 10 months, and in over half of these patients the emergence of the EGFR T790M resistance mutation is detected. Osimertinib is an oral, highly selective, irreversible inhibitor of both EGFR-activating mutations and the T790M-resistance mutation, while sparing the activity of wild-type EGFR. This article reviews clinical trial development of osimertinib in patients with NSCLC, presenting efficacy and safety evidence for its value in the EGFR T790M mutation-positive population and in different settings, including patients with metastatic disease. The preclinical background of clinically acquired resistance to osimertinib is presented and the combination tactics being investigated in an attempt to circumvent this are addressed.
Keywords
Introduction
Over the past decade, the outcomes of biomarker-selected patients with non-small cell lung cancer (NSCLC) have been improved by the in crescendo discovery of activating mutations, with the consequent development of targeted therapies. The first notable success in this personalized medicine era came with the identification of activating mutations in the kinase domain (exons 18–21) of the epidermal growth factor receptor (EGFR) gene, leading to dramatic responses to EGFR tyrosine kinase inhibitors (TKIs). EGFR mutations account for 10–17% of NSCLC cases in North America and Europe and 30–50% of NSCLCs in Asian countries [Kris et al. 2014; Barlesi et al. 2016]. The most common EGFR mutations are the p.Leu858Arg (L858R) point mutation in exon 21 and small in-frame deletions in the region encoded by exon 19, together accounting for approximately 85–90% of all EGFR mutations [Lynch et al. 2004; Paez et al. 2004; Pao et al. 2004]. The first-generation TKIs gefitinib (Iressa®, AstraZeneca Pharmaceuticals, London, United Kingdom) and erlotinib (Tarceva®, F. Hoffmann-La Roche, Basel, Switzerland), and the second-generation TKI afatinib (Giotrif®, Boehringer Ingelheim, Ingelheim, Germany) have shown overall response rates (ORRs) ranging from 50% to 75%, improving progression-free survival (PFS) and quality of life compared with standard platinum-based chemotherapy in patients with EGFR-mutant NSCLC [Mok et al. 2009; Rosell et al. 2012; Yang et al. 2015]. This resulted in their approval as first-line treatments for patients with advanced NSCLC harboring activating mutations in the EGFR kinase domain.
Despite these impressive outcomes, acquired resistance arises after a median period of 9–13 months. Multiple mechanisms have been identified, including secondary mutations in EGFR (notably EGFR T790M), along with mutations in the PIK3CA and BRAF genes and amplifications in ERBB2 and MET [Sequist et al. 2011; Yu et al. 2013; Gainor and Shaw, 2013; Stewart et al. 2015]. The development of a secondary mutation in EGFR when threonine is replaced by methionine at position 790 of exon 20, formally known as T790M (p.Thr790Met), is the most common mechanism, seen in around 50% of cases. While the EGFR-T790M mutation was initially reported as a secondary EGFR resistance mutation, several studies reported de novo EGFR-T790M mutations, sometimes concomitantly with other EGFR-activating mutations [Inukai et al. 2006; Su et al. 2012; Li et al. 2014].
First-generation TKIs compete with adenosine triphosphate (ATP) to bind to the kinase domain of EGFR, and EGFR T790M significantly increases this affinity reducing TKI efficacy [Yun et al. 2008]. Second-generation EGFR TKIs were originally developed to be irreversible EGFR inhibitors with the hope of being active against EGFR-T790M resistance mutations, but they have failed to produce meaningful disease response after resistance to gefitinib or erlotinib [Sequist et al. 2010; Miller et al. 2012; Ellis et al. 2014].
Osimertinib (AZD9291; AstraZeneca Pharmaceuticals), rociletinib (CO-1686; Clovis Oncology, Boulder, United States), olmutinib (BI-1482694/HM61713, Boehringer Ingelheim/Hanmi, Songpa-gu, Korea), ASP8273 (Astellas, Tokyo, Japan), EGF816 (Novartis Pharmaceuticals, Basel, Switzerland), and PF-06747775 (Pfizer, New York, United States) are third-generation EGFR TKIs with selectivity against EGFR-T790M resistance as well as EGFR-sensitizing mutations, all of which have progressed to clinical trials [Cross et al. 2014; Jänne et al. 2015; Sequist et al. 2015b; Lee et al. 2015; Goto et al 2015; Jia et al. 2016]. Table 1 presents available efficacy data from phase I and II clinical trials.
Efficacy of third-generation TKIs in EGFR-T790M-mutated NSCLC populations from phase I and II trials.

Updated results from 2016 ASCO Annual Meeting.
Updated results from phase I TIGER-X trial. While the development of rociletinib was discontinued, the other drugs are still being developed.
bid, twice daily; CI, confidence interval; EGFR, epidermal growth factor receptor; NR, not reached; NSCLC, non-small cell lung cancer; ORR, overall response rate; PFS, progression-free survival; qd, once daily; TKI, tyrosine kinase inhibitor.
To date, osimertinib (Tagrisso™, AstraZeneca Pharmaceuticals) is the only drug to be approved by the European Medicines Agency and the US Food and Drug Administration for treatment of EGFR-T790M mutated NSCLC patients. This review provides an overview of preclinical and clinical data.
Biochemical and preclinical background
Osimertinib is a mono-anilino-pyrimidine compound that acts as a covalent EGFR TKI. In EGFR-recombinant enzyme assays, osimertinib showed potent activity against diverse EGFR mutations (L858R, L858R/T790M, exon 19 deletion, and exon 19 deletion/T790M) and exhibited nearly 200 times greater potency against L858R/T790M than wild-type EGFR. Subsequent murine in vivo studies revealed that osimertinib is metabolized to produce at least two circulating metabolites, AZ5104 and AZ7550.
In biochemical assays, AZ7550 had a comparable potency and selectivity profile to osimertinib, although AZ5104 showed greater potency against exon 19 deletions, T790M mutants (both approximately 8-fold) and wild-type (approximately 15-fold) EGFR [Cross et al. 2014]. In addition, osimertinib and its active metabolites displayed minimal off-target kinase activity for various kinases such as ERBB2/4, ACK1, ALK, BLK, BRK, MLK1, and MNK2 in in vitro studies [Cross et al. 2014]. The area under the plasma concentration–time curve (AUC), maximal plasma concentration (Cmax), and minimal concentration (Cmin) of osimertinib increased over the 20–240 mg dose range with linear pharmacokinetics and the Cmax/Cmin ratio for the 80 mg osimertinib (capsule formulation) was 1.6 [Planchard et al. 2016]. The AUC of osimertinib metabolites AZ5104 and AZ7550 was approximately 10% of osimertinib exposure. Pharmacokinetic exposure was not significantly different between Asian versus non-Asian patients [Planchard et al. 2016]. The median time to Cmax occurred after 6 h (range 3–24). Plasma concentrations decreased with time and the estimated mean half life was 48 h, with clearance (CL/F) of 14.2 (liter/h). Unlike erlotinib, food intake does not impact osimertinib kinetics.
The main metabolic pathways of osimertinib are oxidation (mainly by cytochrome P450, family 3, subfamily A, also known as CYP3A) and dealkylation and it is eliminated primarily in the feces (>65%) and urine (<15%). No clinically significant differences in the pharmacokinetics of osimertinib have been identified in terms of age, sex, ethnicity, body weight, smoking status, mild to moderate renal impairment, or mild hepatic dysfunction. Osimertinib is a competitive inhibitor of CYP3A but does not inhibit CYP2C8, 1A2, 2A6, 2B6, 2C9, 2C19, 2D6, and 2E1. It is a substrate of P glycoprotein and ATP-binding cassette subfamily G member 2, but is not a substrate of organic anion-transporting polypeptide proteins. In a clinical pharmacokinetic study [ClinicalTrials.gov identifier: NCT02163733], the osimertinib exposures were not affected by concurrent administration of omeprazole [Vishwanathan et al. 2015]. Gastric pH modifying agents can be concomitantly used with osimertinib Tagrisso™ without any restrictions.
Clinical efficacy of osimertinib
Phase I clinical trials
The safety and efficacy of osimertinib was assessed in the phase I/II AURA trial [ClinicalTrials.gov identifier: NCT01802632] in patients with locally advanced or metastatic EGFR-mutated NSCLC who had radiologically documented disease progression after treatment with at least one first- or second-generation EGFR TKI [Jänne et al. 2015]. The study included 253 patients who received osimertinib at five dose levels ranging from 20 to 240 mg daily and distributed between two cohorts. Among 31 patients enrolled in the dose-escalation cohort, no dose-limiting toxic effects occurred and an additional 222 patients were treated in five expansion cohorts. All patients had received at least one prior EGFR TKI, and 80% had received prior cytotoxic chemotherapy. The EGFR-T790M mutation was detected in tumors from 138 patients (62%) in the expansion cohort. Of the 253 patients treated across all dose levels, 239 were evaluated for response. The ORR in the combined T790M-positive and T790M-negative populations was 51% [95% confidence interval (CI) 45–58], with 122 patients having a confirmed partial response (PR) and one patient a complete response (CR). Stable disease (SD) was observed in 78 patients (33%) and 34 (14%) experienced disease progression. The disease control rate (DCR; CR, PR or SD) was 84% (95% CI 79–88). The ORR was similar between the 150 Asian and 89 non-Asian patients (50% versus 54%). The 80 mg daily dose was adopted for future studies based on increasing toxicity at 160 and 240 mg daily combined with similar response rates across all dose levels.
Osimertinib exhibited improved efficacy in patients whose tumor harbored the EGFR-T790M mutation. Of 138 patients with a centrally confirmed EGFR-T790M mutation, 127 were evaluable for response. Outcomes were substantially better in this EGFR T790M-postive population compared with patients with T790M-negative tumor, with an ORR of 61% (95% CI 52–70%) versus 21% (95% CI 12–34%), a DCR of 95% (95% CI 90–98%) versus 61% (95% CI 47–73%), and median PFS of 9.6 versus 2.8 months, respectively [Jänne et al. 2015].
Updated results from this trial were recently presented. The efficacy and safety data from the 80 mg expansion cohort in patients with centrally confirmed T790M-positive NSCLC with disease progressing following either one prior therapy with an EGFR-TKI or both an EGFR-TKI and another anticancer therapy, as well as from two expansion cohorts who received osimertinib 80 mg or 160 mg once daily as first-line treatment in patients with EGFR-mutated advanced NSCLC. The former population included 63 patients, 61 of whom were evaluable for response with an ORR of 71% (95% CI 57–82%), a DCR of 93% (95% CI 84–98%), and median PFS of 9.7 (95% CI 8.3-13.6) months [Yang et al. 2016b]. The latter population included 60 patients treated with osimertinib 80 mg (n = 30) or 160 mg (n = 30) daily and all were evaluable. The confirmed ORR was 77% (95% CI 64–87%) with a DCR of 98% (95% CI 89–100%). Median PFS was 19.3 (95% CI 13.7-not calculated), supporting osimertinib use in both first-line and later settings [Ramalingam et al. 2016].
Phase II clinical trials
The 80 mg daily dose was evaluated in the phase II T790M-positive extension cohort of the AURA trial (described above) and in an additional phase II AURA2 study [ClinicalTrials.gov identifier: NCT02094261] designed for patients with confirmed EGFR-mutant T790M-positive locally advanced or metastatic NSCLC who have progressed following prior therapy with an approved EGFR TKI. A preplanned pooled analysis of both studies was recently presented, including a total of 411 patients: 201 patients from the extension cohort of the AURA trial and 210 patients from the AURA2 trial, 397 of whom were included in the response rate evaluation. The ORR was 66% (95% CI 61–71%) and the DCR was 91% (95% CI 88–94%). Median PFS was 11.0 (95% CI 9.6–12.4) months, with a median response duration of 12.5 months (95% CI 11.1–not reached) [Yang et al. 2016b].
Phase III clinical trials
Additional phase III trials involving osimertinib in different settings are ongoing. The phase III First-Line-AURA (FLAURA) trial [ClinicalTrials.gov identifier: NCT02296125] in EGFR-mutated treatment-naïve patients with NSCLC was designed to compare osimertinib 80 mg daily versus current standard of care EGFR TKIs (gefitinib/erlotinib). The AURA3 trial [ClinicalTrials.gov identifier: NCT02151981] was an open-label, randomized study in the second-line setting of osimertinib versus a platinum-based doublet chemotherapy for locally advanced or metastatic NSCLC with the EGFR-T790M mutation. In a very recent press release (dated 18 July 2016) published on the AstraZeneca website, it was announced that the AURA3 phase III trial had met its primary endpoint, demonstrating superior PFS compared with standard platinum-based doublet chemotherapy. In this study that included over 400 patients, osimertinib demonstrated a similar safety profile as in previous trials and results for ORR, DCR, and duration of response were also clinically meaningful compared with chemotherapy. Figure 1 summarizes the development of osimertinib monotherapy from phase I through III trials in patients with advanced EGFR-mutant NSCLC.

Osimertinib development from phase I–III trials in advanced EGFR-mutant NSCLC.
In the adjuvant setting, the ongoing ADjuvant-AURA (ADAURA) trial [ClinicalTrials.gov identifier: NCT02511106] is a double-blind, randomized, placebo-controlled trial assessing the efficacy and safety of osimertinib versus placebo in patients with EGFR-mutated stage IB–IIIA NSCLC following complete tumor resection. The results are not yet available.
Osimertinib in brain and leptomeningeal metastasis
The cumulative incidence of brain metastasis (BM) and leptomeningeal metastasis (LM) in patients with NSCLC is 16–35% and 3–5%, respectively, and is associated with poor prognosis [Schouten et al. 2002; Chamberlain and Kormanik, 1998; Liao et al. 2015]. The real incidence in the EGFR-mutated NSCLC population is unknown, although some data are available from retrospective cohorts reporting an incidence of 24% for BM and 9% for LM [Rangachari et al. 2015; Kuiper et al. 2015]. First- and second-generation EGFR TKIs have limited blood brain barrier penetration [Omuro et al. 2005; Lee et al. 2010; Jamal-Hanjani and Spicer, 2012], with afatinib having the highest efficacy despite its incomplete penetration [Hoffknecht et al. 2015]. Osimertinib induced sustained tumor regression in an EGFR-mutated PC9 mouse BM model and human pharmacokinetics and mouse pharmacokinetics/pharmacodynamics models suggest that doses of 80 mg and 160 mg could be active in human central nervous system (CNS) disease [Kim et al. 2014]. Clinical activity of osimertinib in CNS disease was observed in the phase I AURA trial and an analysis from AURA phase II trials [Ahn et al. 2015] demonstrated the consistent activity of osimertinib in patients with EGFR-mutant T790M NSCLC with and without brain metastases, suggesting its activity in the brain. The analysis of osimertinib pharmacokinetics in cerebrospinal fluid was an exploratory objective in the AURA extension phase II trial.
The phase I BLOOM study [ClinicalTrials.gov identifier: NCT02228369] was designed to assess for the first time the safety, tolerability, pharmacokinetics, and preliminary antitumor activity of AZD3759, an oral EGFR TKI which has excellent CNS penetration and which induces strong regression of BM in a mouse model [Zeng et al. 2015]. In this study, patients with BM and LM may also be enrolled to assess the antitumor efficacy, safety, pharmacokinetics, and potential biological activity of osimertinib 160 mg daily in patients with EGFR-mutated NSCLC whose disease failed to respond to standard treatment and who developed CNS disease (Figure 2). The AZD3759 cohort is ongoing while an update from the EGFR-mutant NSCLC cohort with LM from the osimertinib arm was recently presented; 21 Asian patients with EGFR-mutated NSCLC and LM disease were treated with osimertinib 160 mg daily. All were evaluable for efficacy; seven (33%) had a confirmed radiological response, nine (43%) had stable disease, and neurological function improvement was seen in five (24%) patients [Yang et al. 2016a].

Phase I BLOOM trial to assess the safety, tolerability, pharmacokinetics, and antitumor activity of AZD3759 in EGFR-mutant NSCLC and osimertinib 160 mg daily in EGFR-mutant NSCLC with central nervous system disease. bid, twice daily; BM, brain metastasis; EGFR, epidermal growth factor receptor; NSCLC, non-small cell lung cancer; qd, once daily; TKI, tyrosine kinase inhibitor.
Osimertinib in the first-line setting
Considering the activity of osimertinib against EGFR-sensitizing as well as EGFR-T790M resistance mutations, added to a favorable toxicity profile, in the near future osimertinib may well be considered the option of choice to treat patients with EGFR-mutant NSCLC in the first-line setting. Indeed, preliminary efficacy results are encouraging in patients with EGFR-mutant NSCLC who are treatment naïve as reported from the two expansion cohorts from the phase I AURA trial [Ramalingam et al. 2016]. Results from the phase III FLAURA trial [ClinicalTrials.gov identifier: NCT02296125] are eagerly awaited; if they confirm preliminary results, changes in the current sequence strategy should be discussed.
The safety profile of osimertinib
The dose-limiting toxicity (DLT) of the currently available first- and second-generation TKIs gefitinib, erlotinib, and afatinib is dominated by inhibition of wild-type EGFR in the skin and gastrointestinal tract. Osimertinib exhibited around 200 times greater potency against L858R/T790M than wild-type EGFR, resulting in an attractive EGFR-selective agent in comparison with early-generation TKIs [Cross et al. 2014].
Osimertinib was relatively well tolerated in the phase I AURA trial [Jänne et al. 2015]. No DLT was observed at any dose level up to 240 mg daily. In the combined cohort of 253 patients, the most common adverse events (usually grade 1–2) were diarrhea (47%), skin toxicity (rash/acne, 40%), nausea (22%), and anorexia (21%). Diarrhea and skin toxicity increased with escalating doses of osimertinib. Overall, however, osimertinib was associated with less dermatologic and gastrointestinal toxicity compared with historic data and clinical experience with other approved EGFR TKIs. Only 13% of patients experienced a grade 3 or higher drug-related adverse event. Serious adverse events were observed in 22% of patients (pneumonitis-like events, pulmonary embolism, and pleural effusion), with 6% of patients experiencing a serious drug-related adverse event. Adverse events prompted drug reductions in 7% of patients and drug discontinuation in 6% of patients. The frequency and severity of adverse events were similar between Asian and non-Asian patients. The six cases of potential pneumonitis-like events resolved after treatment discontinuation. Hyperglycemia and QT prolongation were reported in 6 (2%) and 11 (4%) patients, respectively. Among the seven fatal adverse events reported, only one (pneumonia) was considered as possibly drug related.
The phase II AURA extension and the AURA2 trials showed similar results regarding adverse events. The most frequent adverse events (usually grade 1–2) reported from the pooled analysis were rash (41%), diarrhea (38%), dry skin (30%), and paronychia (29%). Grade 3 or higher adverse events were seen in 36% of patients. Any grade interstitial lung disease and QT prolongation were reported in 3% of patients each and only one case of grade 2 hyperglycemia was reported [Yang et al. 2016b]. Unlike osimertinib, hyperglycemia was reported in 36% of patients treated with rociletinib [Sequist et al. 2015b]. Table 2 summarizes drug-related adverse events occurring at the approved dose of 80 mg/day from the phase I AURA trial and the pooled analysis from the AURA extension and AURA2 studies, respectively.
Summary of drug-related adverse events of osimertinib occurring in at least 15% of patients at the approved dose of 80 mg/day from the phase I AURA trial and the pooled analysis of phase II trials (AURA extension and AURA2) in patients with EGFR-T790M-mutant advanced NSCLC.

63 patients with ‘centrally confirmed’ T790M-positive NSCLC who have received osimertinib 80 mg/day.
AE, adverse event; EGFR, epidermal growth factor receptor; ILD, interstitial lung disease; NSCLC, non-small cell lung cancer.
Osimertinib-resistant mutations
Preclinical studies and patient post-progression biopsies allowed identification of multiple resistance mechanisms to first- to third-generation EGFR TKIs. Following the discovery that T790M is the most common acquired resistance mutation to gefitinib and erlotinib, several drugs were developed targeting both EGFR-sensitizing and T790M-resistant mutations. Although various second-generation EGFR TKIs such as afatinib, neratinib, and dacomitinib showed promising activity against T790M-positive cells in preclinical studies, this did not translate into the clinic, with none of them showing efficacy in patients whose disease progressed on the first-generation agents gefitinib and erlotinib [Miller et al. 2012; Reckamp et al. 2014; Sequist et al. 2010]. As a consequence, third-generation EGFR TKIs were developed to target the T790M mutation.
Despite impressive initial outcomes with these new molecules, new mutations and other mechanisms of resistance are emerging. Among these, the C797S mutation in exon 20 of EGFR was found to be the most common mechanism responsible for resistance to osimertinib. This point mutation was identified from circulating tumor DNA (ctDNA) of patients included in the phase I AURA trial whose disease progressed on osimertinib (6 out of 15 patients, 40%) [Thress et al. 2015]. The same mutation was also reported in one case that led to resistance to olmutinib, another oral, third-generation EGFR TKI active against mutant EGFR isoforms, including T790M [Song et al. 2016]. Preclinical EGFR L858R/T790M/C797S mutation cell models exhibited in vitro sensitivity to cetuximab, an antibody that blocks EGFR dimerization [Li et al. 2005; Ercan et al. 2015], but this was not confirmed in in vivo analyses. However, the allosteric inhibitor EAI045 in combination with cetuximab exhibited mechanistic synergy and was effective in mouse models of lung cancer driven by EGFR L858R/T790M and by EGFR L858R/T790M/C797S [Jia et al. 2016]. Interestingly, the allelic context in which C797S was acquired may predict responsiveness to subsequent TKI treatments. For example, if the C797S and T790M mutations are in trans, cells will be resistant to third-generation EGFR TKIs, but sensitive to a combination of first- and third-generation TKIs; and if C797S develops in T790 wild-type cells, this results in resistance to third-generation TKIs, while sensitivity to first-generation TKIs is retained [Niederst et al. 2015]. These data are of great clinical value in sequencing for this mutation in patients with acquired resistance to osimertinib.
The acquired resistance associated with the EGFR T790M mutation can occur either by selection of preexisting EGFR T790M-positive clones or via genetic evolution of initially EGFR T790M-negative drug-tolerant cells, suggesting that cancer cells that survive third-generation TKIs may serve as a key reservoir from which acquired resistance can emerge during treatment [Hata et al. 2016]. Navitoclax (ABT-263, [Ackler et al. (2012)], Abbott Laboratories, Illinois, USA) a BCL-2 family inhibitor, enhances the apoptotic response of late-resistant EGFR T790M cells with decreased sensitivity to EGFR inhibition. The combination of navitoclax with the third-generation EGFR TKI WZ4002 (preclinical compound) induced more apoptosis compared with WZ4002 alone in both in vivo and in vitro analyses. This approach could be an effective strategy for treating EGFR T790M-positive cancers that have a decreased apoptotic response to EGFR inhibition [Hata et al. 2016]. An ongoing phase Ib trial is evaluating the safety and tolerability of the osimertinib/navitoclax combination in patients with EGFR-mutant NSCLC following resistance to prior EGFR TKIs [ClinicalTrials.gov identifier: NCT02520778].
Additional EGFR-independent mechanisms of resistance to osimertinib have been reported. NRAS mutations, including a novel E63K mutation, and amplifications of wild-type NRAS or KRAS have been described as mechanisms of acquired resistance to osimertinib but also to gefitinib and afatinib [Eberlein et al. 2015]. In vitro, a combination of osimertinib with the MEK inhibitor selumetinib prevented emergence of resistance in PC9 (Ex19del) cells and delayed resistance in NCI-H1975 (L858R/T790M) cells. In vivo, concomitant osimertinib with selumetinib caused regression of osimertinib-resistant tumors in an EGFR-mutant/T790M transgenic model [Eberlein et al. 2015]. This association is been evaluated in the phase Ib TATTON trial [ClinicalTrials.gov identifier: NCT02143466]. In addition, the combination of trametinib, another MEK inhibitor, with WZ4002 prevents the development of acquired resistance in EGFR-mutant lung cancer models [Tricker et al. 2015].
Amplifications in HER2 and MET genes were also described as potential mechanisms of acquired resistance to osimertinib in patients with EGFR-T790M-mutant NSCLC [Planchard et al. 2015]. Additionally, loss of T790M at the time of progression may be mediated by overgrowth of cells harboring HER2 amplification, BRAF V600E or PIK3CA mutations, as was recently reported following examination of plasma specimens from patients included in the phase I AURA trial [Oxnard et al. 2015].
In addition, resistant tumors have been reported to show phenotypic changes, such as small-cell lung cancer transformation or epithelial to mesenchymal transition [Sequist et al. 2011; Yu et al. 2013; Kim et al. 2015]. Figure 3 summarizes the known mechanisms of resistance to third-generation EGFR TKIs.
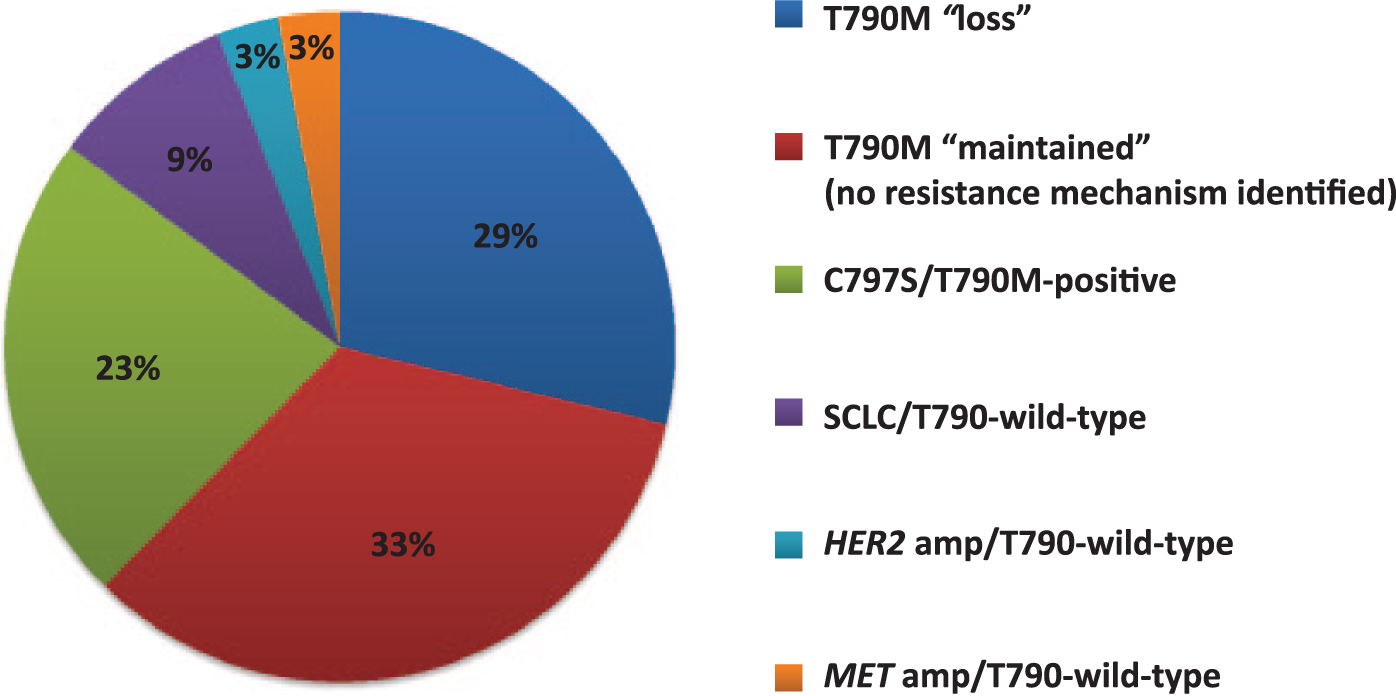
Mechanisms of resistance to third-generation EGFR TKIs osimertinib and rociletinib. Data from Piotrowska et al. [2015], Thress et al. [2015], Yu et al. [2015], Planchard et al. [2015], and Kim et al. [2015]. amp, amplification; EGFR, epidermal growth factor receptor; SCLC, small cell lung cancer; TKI, tyrosine kinase inhibitor.
Overcoming osimertinib-resistant disease
The heterogeneity in the acquired resistance mechanisms to osimertinib provides the basis for investigating different inhibitory combination strategies. Therefore, osimertinib-based combinations are currently being investigated in several studies. The multiarm phase Ib TATTON trial [ClinicalTrials.gov identifier: NCT02143466] was designed to evaluate the safety, tolerability, and preliminary antitumor activity of osimertinib in combination with durvalumab (anti-PD-L1 monoclonal antibody), savolitinib (MET inhibitor) or selumetinib (MEK 1/2 inhibitor) in patients with advanced EGFR-mutant NSCLC whose disease has progressed on an EGFR TKI. Preliminary results from the osimertinib/durvalumab arm were recently presented [Ahn et al. 2016]. In patients with prior EGFR TKI therapy, investigator-assessed ORR was 67% (6/9) in those with T790M-mutant tumors compared with 21% (3/14) in T790M-negative NSCLC. Regarding safety data, interstitial lung disease was reported in 38% (13/34) of patients, higher than would be expected with either drug alone. Five events were grade 3–4 and there were no fatalities; most cases were managed using steroids [Ahn et al. 2016]. Based on these data, the recruitment into the osimertinib plus durvalumab arm of TATTON is currently on hold, but expansion cohorts of the MET and MEK inhibitor combinations are ongoing. In addition, the phase III Combination-AURA in Lung (CAURAL) trial [ClinicalTrials.gov identifier: NCT02454933] is being conducted in second-line metastatic EGFR-mutant/T790M-positive NSCLC patients testing osimertinib plus durvalumab versus osimertinib monotherapy for their impact on PFS. This study was also stopped prematurely due to the pulmonary toxicity observed in the TATTON trial.
On the basis of preclinical observations that afatinib (an irreversible ErbB family blocker) plus cetuximab (an anti-EGFR monoclonal antibody) overcame T790M-mediated resistance [Regales et al. 2009], the combination was evaluated in a phase Ib trial enrolling 126 heavily pretreated patients with advanced EGFR-mutant NSCLC who developed resistance to first-generation erlotinib/gefitinib. The ORR was 29%, comparable in both T790M-positive and T790M-negative tumors (32% versus 25%) and the median PFS was 4.7 (95% CI 4.3–6.4) months [Janjigian et al. 2014]. However, dual EGFR inhibition significantly improves toxicity, including (all grade) rash (seen in 90% of patients), diarrhea (71%), and stomatitis (56%). Grade 3–4 adverse events were observed in 46% of patients [Janjigian et al. 2014]. A randomized phase II/III trial [ClinicalTrials.gov identifier: NCT02438722] of afatinib plus cetuximab versus afatinib alone is currently open in treatment-naïve patients with advanced EGFR-mutant NSCLC. The dual EGFR blockage is being evaluated in a phase I trial [ClinicalTrials.gov identifier: NCT02496663] combining osimertinib with the anti-EGFR monoclonal antibody necitumumab to assess safety and determine the optimal dose in patients with EGFR-mutant advanced NSCLC whose disease has progressed on a previous EGFR TKI.
The dual vascular endothelial growth factor receptor (VEGFR) and EGFR blockade inhibits tumor growth in EGFR TKI resistance xenograft models [Naumov et al. 2009]. Indeed, this hypothesis was confirmed in two phase II clinical trials in patients with EGFR-mutant NSCLC who are treatment naïve: the randomized Japanese (JO25567) trial comparing erlotinib plus bevacizumab versus erlotinib alone, and the single-arm (Bevacizumab and ErLotinib In EGFR Mut+ NSCLC [BELIEF]) trial in white patients. Median PFS was encouraging and similar in both studies, supporting the combination in the first-line setting [Seto et al. 2014; Stahel et al. 2015]. A phase I trial was thus designed to evaluate the safety of two osimertinib-based combination strategies, with necitumumab or ramucirumab (an anti-VEGFR2 monoclonal antibody) in patients with advanced EGFR-T790M-mutant NSCLC after progression on first-line EGFR TKI therapy [ClinicalTrials.gov identifier: NCT02789345]. Finally, the combination of osimertinib and bevacizumab will be evaluated in another phase I/II 3+3 dose-escalation design [ClinicalTrials.gov identifier: NCT02803203] to test the safety of combining these drugs.
For patients whose tumors undergo small-cell lung cancer transformation, platinum-based plus etoposide chemotherapy is recommended. Table 3 provides information about ongoing and forthcoming osimertinib-based combination trials to treat or prevent osimertinib-acquired resistance.
Ongoing and forthcoming osimertinib-based combination trials.

MTD, maximal tolerated dose; EGFR, epidermal growth factor receptor; NSCLC, non-small cell lung cancer; ORR, overall response rate; PFS, progression-free survival; TKI, tyrosine kinase inhibitor; VEGFR, vascular endothelial growth factor receptor.
Osimertinib in the era of liquid biopsies
To date, there is increasing evidence that a single tissue biopsy may not adequately represent intrinsic tumor heterogeneity, particularly in cases of disease progression. Moreover, tumor location and the risk of complications are limitations for new tissue biopsies. Emerging evidence suggests that analysis of ctDNA could more broadly capture the spectrum of resistant clones that may appear throughout the course of the disease. Performing serial ctDNA analyses could also evaluate the longitudinal response, and potentially detect resistance mutations before documented radiographic progression [Thress et al. 2015; Piotrowska et al. 2015]. For example, ctDNA was used to detect T790M in plasma in 70% (23 of 35) of patients treated with rociletinib who had a T790 wild-type tissue biopsy [Sequist et al. 2015a]. Notably, the efficacy of rociletinib was equivalent whether T790M was detected in tissue or in plasma, suggesting that noninvasive testing may be adequate for predicting response and could provide additional information in patients with tissue biopsies which are negative for T790M [Thress et al. 2015; Piotrowska et al. 2015]. In addition, early acquisition of EGFR-resistance mutations could be found by measuring ctDNA in the urine [Husain et al. 2015]. Recently, genotype-matched results from plasma, tissue, and urine samples from patients included in the phase I/II TIGER-X trial were reported. Considering the tissue sample as the reference, sensitivity for detecting T790M mutation in plasma and urine was 80.9% and 81.1%, respectively. Response rates were similar in the T790M-mutant population irrespective of whether the status was identified in plasma, tissue, or urine [Wakelee et al. 2016].
Plasma samples from 192 patients enrolled in the phase I AURA trial were collected and genotyped. Sensitivity for detecting EGFR-sensitive and T790M-resistant mutations was 87% and 78%, respectively. Clinical response rates were greater in T790M-positive patients, as assessed by either tissue or plasma genotyping [Thress et al. 2014]. Eligibility for treatment with osimertinib will be dependent on mutational status, which will be determined via a validated diagnostic test based on a tumor tissue sample or plasma. Availability of a blood-based test for ctDNA means that physicians and patients have multiple options to test for a T790M-resistant mutation.
Discussion
The EGFR-T790M mutation is the main mechanism of acquired resistance to first- and second-generation EGFR TKIs and represents a barrier in the treatment of patients with EGFR-mutant advanced NSCLC. Osimertinib has demonstrated strong efficacy and safety data in phase I and II trials, and has become the first EGFR inhibitor approved for the treatment of NSCLC with the EGFR-T790M mutation. Patients with advanced NSCLC with EGFR-activating mutations whose disease progresses on a first-line EGFR TKI have traditionally been offered platinum-doublet chemotherapy as second-line treatment. Platinum-doublet chemotherapy shows ORRs of approximately 30%, slightly higher than the rate observed in the T790M-negative population, but significantly lower than the 61–71% ORR reported in T790M-positive cohorts in phase I and II trials with osimertinib. The phase III AURA3 trial [ClinicalTrials.gov identifier: NCT02151981] confirms the superiority of osimertinib for treating patients with EGFR-T790M-mutant NSCLC in the second-line setting compared with standard pemetrexed-containing/platinum-based chemotherapy. In addition, considering the favorable safety profile of osimertinib added to its systemic and CNS efficacy, osimertinib is currently the most attractive option in the second-line setting for patients with T790M-mutant NSCLC, delaying chemotherapy to the third-line setting, as well as for patients with T790M-postive NSCLC with brain or leptomeningeal metastases. Figure 4 illustrates a potential treatment algorithm for patients with EGFR-mutated advanced NSCLC. If we take into consideration the encouraging response outcomes (ORR 77%, DCR 98%) and PFS (approximately 19 months in the first-line setting), osimertinib is likely to be the best option for treating patients with advanced EGFR-mutant NSCLC as first-line therapy. The phase III FLAURA trial [ClinicalTrials.gov identifier: NCT02296125] probably gives us the approach for better positioning osimertinib regarding current EGFR TKIs in order to improve sequences with the final objective of improving patient outcomes.

Potential treatment algorithm for patients with EGFR-mutated advanced NSCLC. CT, chemotherapy; EGFR, epidermal growth factor receptor; MoR, mechanism of resistance; mPFS, median progression-free survival; ORR, overall response rate; qd, once daily; SCLC, small cell lung cancer; TKI, tyrosine kinase inhibitor.
The role of EGFR TKIs in the adjuvant setting for nonmetastatic EGFR-mutated lung cancer is in a very early development stage and remains controversial. Erlotinib and gefitinib were evaluated in prospective trials suggesting an improvement in disease-free survival, but none of these trials demonstrate a benefit in overall survival [Goss et al. 2013; Janjigian et al. 2011; Pennell et al. 2014; Kelly et al. 2015]. The phase III ADAURA trial [ClinicalTrials.gov identifier: NCT02511106] comparing osimertinib with placebo as adjuvant therapy in stage IB-IIIA EGFR-mutated NSCLC following complete tumor resection is currently recruiting patients, and the jury remains out until at least preliminary results become available. These studies have the potential to significantly expand the role of osimertinib in the treatment algorithm for EGFR-mutated NSCLC.
The heterogeneity of resistant cancers plays an important role, not only in terms of response and resistance to the new EGFR TKIs, but in allowing different combination strategies to be more effective in preventing and delaying resistance mechanisms. Due to its safety profile, osimertinib is now considered an attractive drug to combine with other targeted therapies. While combinations with MEK and MET inhibitors as well as antiangiogenic agents are promising, we must exercise precaution with respect to their toxicity profiles. Table 3 summarizes ongoing and forthcoming osimertinib-based combination trials.
Conclusion
Osimertinib, developed in less than 3 years, represents one of the fastest cancer drug development programs with respect to obtaining approval for the treatment of patients with EGFR-T790M NSCLC whose disease has progressed on EGFR TKIs. The encouraging results obtained in patients with EGFR-mutant NSCLC in the first-line setting place it as an established critical drug in this scenario.
Footnotes
Acknowledgements
The authors thank Sarah MacKenzie PhD for English editing.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of interest statement
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.