
Abstract
The discovery of epidermal growth factor receptor activating mutations (EGFR Mut+) has determined a paradigm shift in the treatment of non-small cell lung cancer (NSCLC). In several phase III studies, patients with NSCLC EGFR Mut+ achieved a significantly better progression-free survival when treated with a first- (gefitinib, erlotinib) or second-generation (afatinib) EGFR tyrosine kinase inhibitor (TKI) compared with standard chemotherapy. However, despite these impressive results, most patients with NSCLC EGFR Mut+ develop acquired resistance to TKIs. This review will discuss both the mechanisms of resistance to TKIs and the therapeutic strategies to overcome resistance, including emerging data on third-generation TKIs.
Keywords
Introduction
The introduction into clinical practice of targeted therapies for molecularly defined tumors has determined a paradigm shift in cancer treatment. In non-small cell lung cancer (NSCLC), epidermal growth factor receptor activating mutations (EGFR Mut+), the most common being exon 19 deletions (~60%) and L858R substitution in exon 21 (~30%), identify a subtype of lung cancer whose growth is dependent on EGFR pathway activation [Lynch et al. 2004; Rosell et al. 2009]. The prevalence of these mutations varies by ethnicity: they have been found in around 15% of white populations, in around 40% of East Asians and in around 60% of selected Asian patients (female sex, never/light smoker, adenocarcinoma). It has been also demonstrated that EGFR Mut+ led to increased affinity of EGFR tyrosine kinase inhibitors (TKIs) for the mutant receptor, thus conferring sensitivity to TKI treatment. First-generation TKIs, such as gefitinib and erlotinib, act as competitive reversible inhibitors of adenosine triphosphate (ATP), therefore preventing the autophosphorylation of the TK domain, and blocking the activation of the EGFR downstream pathway. Second-generation TKIs, including the recently approved afatinib, irreversibly alkylate cysteine 797 in the ATP binding site and interact with other members of the HER family [Keating et al. 2014]. In several phase III studies, patients with EGFR Mut+ NSCLC achieved a significantly better progression-free survival (PFS) when treated with a first- or second-generation TKI compared with standard chemotherapy [Mok et al. 2009; Han et al. 2012; Mitsudomi et al. 2010; Maemondo et al. 2010; Zhou et al. 2011; Rosell et al. 2012; Sequist et al. 2013; Wu et al. 2014] (Table 1). In these studies no significant difference was observed in terms of overall survival (OS) due to the high percentage of patients receiving chemotherapy who crossed to TKIs at disease progression. However, the recently published analysis of OS data from two randomized trials (LUX-Lung 3 and LUX-Lung 6) demonstrated that, although afatinib did not improve OS in the whole population of either trial, OS was significantly improved for patients with EGFR exon 19 deletion [Yang et al. 2015]. In addition, the majority of the phase III randomized studies, in which the patient-reported outcome assessment was evaluated, showed a significant advantage in favor of patients treated with a TKI [Yang et al. 2013]. In view of the reported data, TKIs have become the standard of care in patients with NSCLC EGFR Mut+ [Reck et al. 2014]. However, despite a good initial response, the occurrence of acquired resistance limits the long-term efficacy of these novel agents.
Randomized studies of first-line EGFR TKIs in patients with NSCLC EGFR Mut+.

Subgroup of patients with EGFR Mut+.
First phase III study including Western patients.
EGFR Mut+, epidermal growth factor receptor activating mutation; NSCLC, non-small cell lung cancer; PFS, progression-free survival; RR, Response Rate; TKI, tyrosine kinase inhibitor; CHT, Chemotherapy; CBDCA, Carboplatin; CDDP, Cisplatin; TAX, Taxol; TXT, Docetaxel; PEM=,Pemetrexed; GEM, Gemcitabine.
The current review focuses on the main resistance mechanisms to TKIs and analyzes the possible strategies to overcome resistance.
Mechanisms of resistance to EGFR TKIs
Usually we distinguish a primary or an acquired resistance to TKIs: patients with primary resistance are initially refractory to treatment, whereas acquired resistance may occur in patients initially responding to TKIs. Jackman and colleagues [Jackman et al. 2010] proposed a set of criteria to define the acquired resistance to TKIs: previous treatment with a TKI; either of the following: evidence of benefit from treatment with a TKI, progression of disease while on treatment with gefitinib or erlotinib within the last 30 days and no intervening systemic therapy between cessation of gefitinib or erlotinib and initiation of new therapy. Camidge and colleagues distinguish two types of acquired resistance to TKIs: a pharmacological and a biological resistance [Camidge et al. 2014]. Pharmacological resistance indicates cancer progression caused by inadequate drug exposure against the target protein; instead, biological resistance reflects the evolution of cancer cells in the presence of adequate drug exposure. In addition, it is possible to distinguish EGFR-dependent or EGFR-independent resistance mechanisms (Table 2). Several studies have identified some of the mechanisms involved in the development of acquired resistance to TKIs, although in up to 30% of cases they are still unexplained. Results come from the molecular analysis performed in patients receiving TKI therapy, who underwent tumor biopsy at recurrence. For example, Sequist and colleagues performed a genetic and histological analysis in 37 patients with NSCLC with acquired resistance to TKIs, who underwent multiple repeated biopsies over the course of their treatment [Sequist et al. 2011]. The repeated tumor biopsies revealed the presence of T790M mutation (49%), EGFR T790M amplification (8%), MET amplification (5%), PIK3CA mutations (5%) and conversion from NSCLC to small-cell lung cancer (14%). Considering the difficulty in obtain tissue specimens during disease progression, several studies investigated the potential of noninvasive methods, so-called ‘liquid biopsies’, such as analysis of circulating tumor cells (CTCs) or plasma cell-free DNA (cfDNA) [Maheswaran et al. 2008; Oxnard et al. 2014]. Maheswaran and colleagues isolated CTCs from patients with metastatic NSCLC, then performed EGFR mutational analysis on DNA recovered from CTCs and compared the results with those from concurrently isolated cfDNA and from the original tumor specimens [Maheswaran et al. 2008]. In this study, molecular analyses showed the expected EGFR activating mutations in CTCs in 11 out of 12 patients (92%) and in cfDNA in 4 out of 12 patients (33%). Oxnard and colleagues demonstrated the presence of EGFR Mut+ in cfDNA at diagnosis in patients with EGFR Mut+ NSCLC, its reduction or disappearance during therapy with erlotinib, and its subsequent reemergence along with the drug resistance EGFR T790M mutation [Oxnard et al. 2014].
Main mechanisms involved in resistance to EGFR TKIs.

CRKL, v-crk sarcoma virus CT10 oncogene homolog avian-like; EGFR, epidermal growth factor receptor; EMT, epithelial to mesenchymal transition; HER2, human epidermal growth factor receptor 2; HGF, hepatocyte growth factor; MET, tyrosine kinase receptor for hepatocyte growth factor; TKI, tyrosine kinase inhibitor.
Overall, the two most common acquired resistances to TKIs are represented by T790M mutation and MET amplification, occurring in approximately 60% of cases. The presence of T790M, which is found in approximately 50% of patients with TKI resistance, altering the conformation of the TK domain of the EGFR restores the affinity of the receptor for ATP, thus reducing the ability of TKIs to compete with ATP [Yun et al. 2008]. T790M is analogous to the ABL T3151 KIT T670I gatekeeper mutations observed in imatinib-resistant chronic myeloid leukemia (CML) and gastrointestinal stromal tumor (GIST), respectively [Oxnard et al. 2011]. Chmielecki and colleagues developed isogenic TKI-sensitive and TKI-resistant pairs of cell lines that mimic the behavior of human tumors [Chmielecki et al. 2011]. They demonstrated that the drug-sensitive and drug-resistant EGFR Mut+ cells exhibited differential growth kinetics, with the drug-resistant cells showing slower growth.
MET is a transmembrane TK receptor that binds to the hepatocyte growth factor (HGF). MET amplification confers resistance through ERBB3-mediated activation of downstream PI3K/AKT signaling, bypassing the inhibited EGFR [Engelman et al. 2007]. Activation of MET through its ligand HGF, produced by stromal as well as tumor cells, may also promote resistance to TKIs by restoring the PI3K/AKT signaling pathway via phosphorylation of MET through GAB1 signaling, but not EGFR or ERBB3 [Yano et al. 2008]. Although MET amplification can occur with the EGFR T790M mutation, about 60% of MET amplification is found without T790M mutation. The T790M mutation may also exist before treatment; similarly, MET amplification has been reported in EGFR Mut+ tumors before TKI exposure [Rosell et al. 2011; Turke et al. 2010].
Other mechanisms of acquired resistance to TKIs include HGF overexpression, Human Epidermal Growth factor Receptor 2 (HER2) amplification, epithelial mesenchymal transition, AXL activation, CRKL (v-crk sarcoma virus CT10 oncogene homolog avian-like) amplification, PTEN (phosphatase and tensine homolog) downregulation, FAS-NFκB (apoptosis stimulating fragment nuclear factor κB) activation and conversion to small-cell lung cancer [Cheung et al. 2011; Yamamoto et al. 2010; Suda et al. 2012].
Strategies to treat patients with NSCLC with EGFR TKI resistance and to overcome resistance
In clinical practice no standard treatment approach exists to overcome TKI resistance. Gandara and colleagues suggest distinguishing a central nervous system progressive disease (PD), an oligo PD and a systemic PD [Gandara et al. 2014]. The authors reported that in patients with central nervous system PD or oligo PD, a local-regional approach plus continuing TKI therapy could be a rational option. Moreover, preclinical and clinical data showed that the clones carrying EGFR T790M proliferate slower than the mutated EGFR without T790M, therefore giving a rationale for continuing TKIs when a local recurrence occurs, and treating the site of progression with a local therapy [Chmielecki et al. 2011; Li et al. 2014b; Hata et al. 2013; Oxnard et al. 2011]. In the study by Li and colleagues, including 41 patients treated with TKIs beyond progression, a significantly longer Progression Free Survival 2 (PFS2) (6.3 versus 2.6 months, p = 0.002) and OS (39.8 versus 23.2 months, p = 0.044) were observed in patients with T790M mutation compared with those without [Li et al. 2014b]. Considering that T790M mutation represents the most frequent acquired resistance mechanism, researchers developed specific drugs to target this mutation. Currently three agents AZD9291, CO-16886 and HM61713 are under study for the treatment of patients with T790M+ NSCLC. AZD9291 is an irreversible inhibitor of EGFR and T790M mutations with a reduced affinity for wild-type EGFR [Ward et al. 2013]. Patients with EGFR Mut+ NSCLC and acquired resistance to TKIs were enrolled into a phase I trial [Yang et al. 2014]. A total of 31 and 201 patients were enrolled in the escalation and expansion cohort, respectively. The primary endpoint was safety, but preliminary efficacy was included as a secondary endpoint. The investigators found that AZD9291 was well tolerated, with no dose-limiting toxicities (DLTs) reported and the maximum tolerated dose (MTD) not reached. Similarly to other TKIs, the most common adverse events (AEs) observed were diarrhea (39%), rash (36%) and nausea (18%). AZD9291 was more beneficial in patients with T790M+ disease, with an overall response rate (ORR) of 61% and durable responses of over 6 months versus an ORR of 21% in patients with T790M– disease [Steuer et al. 2014]. In the overall population an ORR of 53% was observed. Based on these data, in April 2014 AZD9291 was granted breakthrough therapy designation by the US Food and Drug Administration (FDA) for the treatment of patients with NSCLC and EGFR T790M+ whose disease has progressed during treatment with a TKI. A new drug may be designated as a breakthrough therapy by the FDA if it is intended to treat a serious or life-threatening disease and preliminary clinical evidence suggests it provides a substantial improvement over existing therapies. Once the breakthrough therapy designation is obtained, the FDA and sponsor work together to determine the most efficient path forward. Several trials with AZD9291 in patients with NSCLC T790M+ are ongoing with a once daily 80 mg dose (Table 3). The ongoing phase III FLAURA trial [ClinicalTrials.gov identifier: NCT02296125] will assess the efficacy and safety of AZD9291 versus gefitinib or erlotinib as first-line treatment in patients with advanced NSCLC EGFR Mut+. CO-1686 (rociletinib) is an irreversible orally delivered third-generation TKI that specifically targets the mutant form of EGFR, including T790M [Walter et al. 2013]. A phase I/II study (TIGER X Trial) enrolled patients with advanced EGFR Mut+ NSCLC pretreated with TKIs; T790M+ status was a requirement for the phase II cohort [Sequist et al. 2014]. In this trial no MTD was identified and the most common AE noted was hyperglycemia (grade 3 in 22% of the patients). In terms of efficacy, among the patients with confirmed T790M+ the ORR was 58%. Ongoing trials with CO-1686 are listed in Table 3. As with AZD9291, in May 2014 the FDA granted breakthrough therapy designation for CO-1686 as treatment for patients with NSCLC T790M+ after progression on a prior TKI. HM61713 is another orally administered third-generation TKI. HM61713 was studied in a phase I Korean study enrolling patients with EGFR Mut+ NSCLC that had progressed on prior TKI therapy. Preliminary results showed that HM61713 is a safe and effective treatment, especially in patients with T790M mutation [Kim et al. 2014].
Clinical trials with third-generation TKIs.
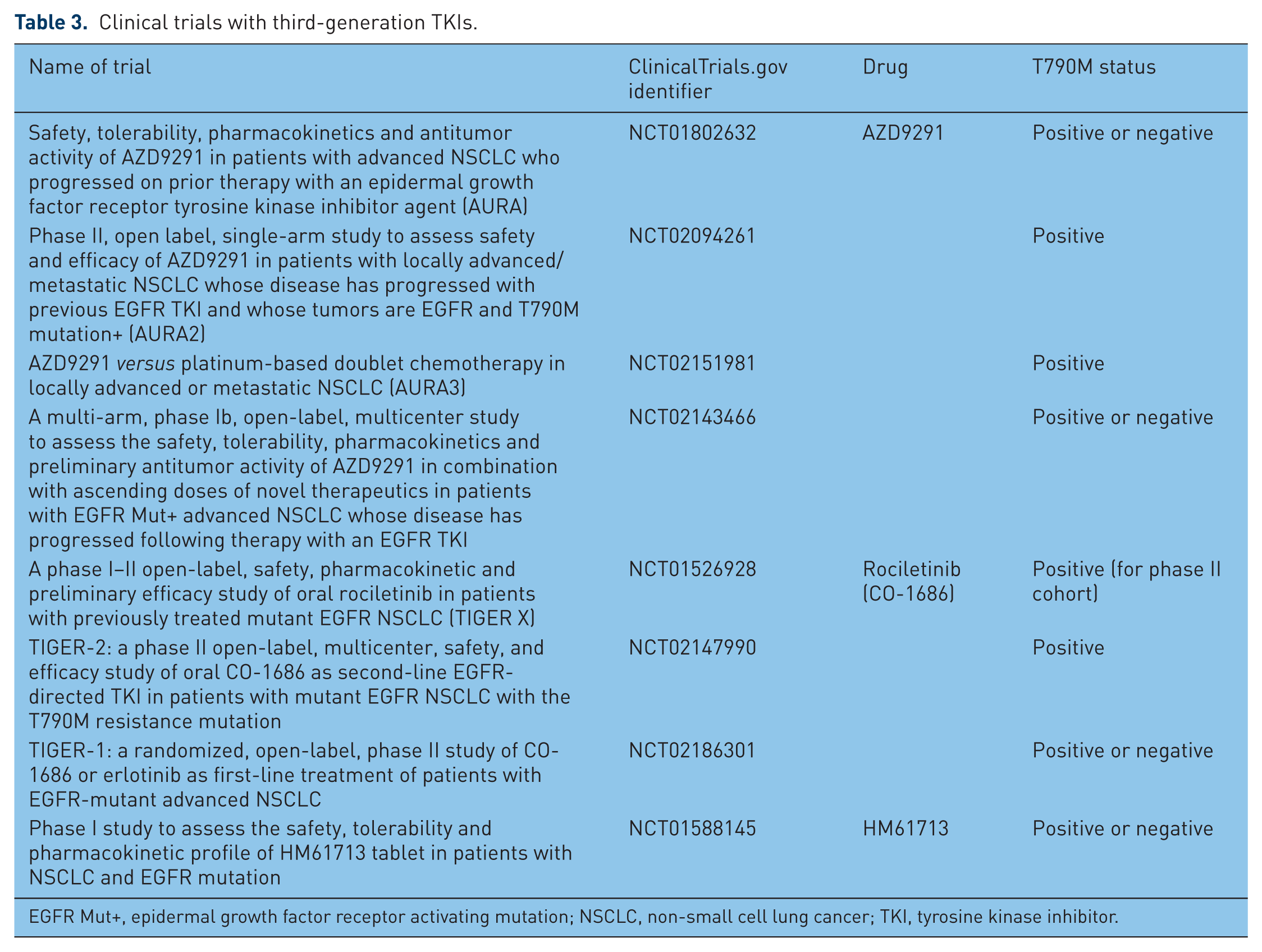
EGFR Mut+, epidermal growth factor receptor activating mutation; NSCLC, non-small cell lung cancer; TKI, tyrosine kinase inhibitor.
An alternative clinical strategy is to give the patient a rechallenge with a TKI after a period of chemotherapy treatment: without the selected pressure of TKIs, previously arrested TKI-sensitive cells can repopulate more quickly than TKI-resistant cells, thus resulting in an efficient inhibition by the reintroduction of TKIs. In the retrospective study by Hata and colleagues, 59 (76%) of the 78 patients underwent TKI rechallenge: median PFS of patients with T790M was 5.0 months and without T790M 2.7 months [Hata et al. 2013]. The authors concluded that TKI rechallenge may be effective after initial TKI failure, particularly in patients with T790M mutation. The ICARUS study [ClinicalTrials.gov identifier: NCT01530334] is an Italian phase II single-arm trial that evaluated the activity of gefitinib as treatment rechallenge in patients with advanced EGFR Mut+ NSCLC who previously responded to gefitinib and received subsequent chemotherapy. The study has been completed and the results will be available soon. Table 4 summarizes the main possible clinical approaches (switch therapy, continue same TKI, add therapy to TKI) to treat patients with NSCLC and TKI acquired resistance. Some of these approaches have been evaluated in clinical studies. In the IMPRESS trial patients with locally advanced or metastatic EGFR Mut+ NSCLC, prior progression disease (PD) on first line gefitinib and no previous chemotherapy were randomized to begin chemotherapy with cisplatin and pemetrexed or to continue gefitinib with the same chemotherapy [Mok et al. 2014] (Figure 1). There was no statistically significant improvement in PFS, the primary endpoint of the study, for gefitinib versus placebo. This phase III study indicates that in patients with disease progressing on first-line TKIs, if we decide to use chemotherapy it is advisable to suspend the TKI. In the ASPIRATION trial, patients with EGFR Mut+ lung cancer were treated with erlotinib, and at the time of RECIST progression, their physicians decided whether to continue or stop erlotinib [Park et al. 2014]. In this study the first PFS (defined by the RECIST criteria) was 11 months, while the second PFS (defined by the physician’s discretion to stop the drug) was 14.1 months, then with a gain of 3.1 months for patients who received treatment beyond progression. The ASPIRATION trial showed that in clinically asymptomatic patients, instead of switching to another therapy just because of radiologic progression, to maintain the TKI as long as possible is a viable treatment option. The authors declared that this approach is a reflection of a real-life situation, in which we do not use only the RECIST criteria as a decision point. The current National Comprehensive Cancer Center guidelines indicate that the decision to switch is based on the site of metastasis, whether the patient is symptomatic and how well the drug is tolerated [National Comprehensive Cancer Center, 2015]. On the basis of preclinical observations showing a synergistic effect, Janjigian and colleagues conducted a study to investigate the safety and efficacy of combined EGFR blockade with afatinib and cetuximab in patients with EGFR Mut+ that had acquired resistance to erlotinib or gefitinib [Janjigian et al. 2014]. In this study a total of 37 patients (29%) reported an objective response rate, including 22 (18%) who had tumor shrinkage of 50% or more; the median duration of response to the combination therapy was 5.7 months. In addition, the objective response rate was comparable in T790M+ and T790M– tumors (32% versus 25%; p = 0.341). Grade 3 and 4 treatment-related AEs were noted in 44% and 2% of patients, respectively, and the most common grade 3 events were rash (20%) and diarrhea (6%).
Main possible approaches to treat patients with NSCLC and TKI-acquired resistance.

NSCLC, non-small cell lung cancer; TKI, tyrosine kinase inhibitor.

IMPRESS trial. EGFR, epidermal growth factor receptor.
Other strategies to overcome the resistance mechanisms to TKIs are actually under study. For example, Huang and colleagues reported that the loss of MED12, a component of the transcriptional mediator complex, is involved in the development of resistance to targeted therapies in cancer lines with activating EGFR or BRAF mutations or EML4-ALK rearrangement [Huang et al. 2012]. The authors found that the inhibition of transforming growth factor β receptor signaling restored drug responsiveness in MED 12 deficient cells, suggesting a strategy to treat drug-resistant tumors that have lost MED12.
Japanese researchers showed that a combination of erlotinib and bevacizumab partially reverses resistance to erlotinib in xenograft models with high levels of Vascular Endotelial Growth Factor (VEGF) protein [Li et al. 2014a]. These results suggest that addition of bevacizumab could enhance antitumor activity of erlotinib and partially reverse resistance to TKIs, by increasing erlotinib concentration in tumors that express high levels of VEGF protein. More recently, Nakade and colleagues evaluated the effect of triple inhibition of EGFR, Met and angiogenesis on HGF triggered EGFR TKI resistance in EGFR-mutant lung cancer [Nakade et al. 2014]. The authors demonstrated that triple inhibition of EGFR, HGF/Met and VEGF/VEGF receptor 2, by either a triplet of clinical drugs or TAS-115, a novel dual TKI for Met and VEGF receptor 2, combined with erlotinib, may be useful for controlling progression of EGFR-mutant cancer.
Another potential solution to overcome multiple mechanism of resistance in patients with NSCLC could be to target ROR1, a pseudokinase that mediates the balance between survival and apoptosis [Chong and Janne, 2013].
Conclusion
In patients with NSCLC EGFR Mut+ the development of drug resistance causes progression of disease in spite of a good initial response to TKIs. In more than half of these patients, the treatment resistance results from T790M mutation. In the near future the availability of agents targeting T790M will likely dictate the need to change clinical practice standards, including the importance of the rebiopsy at progression. Anyway, there are no fixed rules to switch to a second-line therapy in patients with acquired resistance to TKIs; in fact, many patients with slow (>6 months of partial response/stable disease), asymptomatic minimal PD or new brain metastasis controlled locally can be continued on their original TKI and switched to another treatment when the physician feels that the clinical benefit can no longer be maintained [Gandara et al. 2014].
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.