
Abstract
A number of therapeutic agents are available for the treatment of asthma, including inhaled corticosteroids, long- and short-acting beta-agonists, leukotriene-modifying agents, long- and short-acting anticholinergic agents, chromones, theophylline, allergen immunotherapy, and oral corticosteroid therapy. All available therapies, despite their proven efficacy, are purely symptomatic including the topical steroids. This issue has led to the development of several biologic agents to aid in asthma management and to potentially alter the course of the disease by interfering with specific aspects of inflammation which may modify remodeling in the airways. Monoclonal antibodies have offered a class of therapeutic agents that enhance treatment options for patients with moderate-to-severe persistent asthma. As such, this article provides an overview of present and future monoclonal antibody therapies for the treatment of patients with severe asthma.
Antibody structure, function and the production of monoclonal antibodies
Antibody structure and function
Antibodies, or immunoglobulins, are a family of structurally related glycoproteins produced by B cells. An individual antibody molecule has a core consisting of two light chains and two heavy chains. The heavy chains and light chains are composed of amino-terminal variable (V) regions and carboxy-terminal constant (C) regions. Most of the effector functions of antibodies are mediated by the C regions on heavy chains [Abbas, 2010].
Human antibodies have been divided into five isotypes based on their heavy chain constant regions. The five groups consist of immunoglobulins IgG, IgA, IgM, IgD, and IgE; which can be subdivided based upon two distinct functional units including: the fragment that mediates antigen binding (Fab) and the so-called crystallizable fragment (Fc). The Fab unit contains the variable region, which is composed of three hypervariable complementarity-determining regions (CDRs). CDRs are involved with antigen specificity and form the binding site for antigen attachment [Weiner et al. 2010]. This has been reviewed extensively elsewhere.
Production of monoclonal antibodies
Monoclonal antibodies (mAb) to human targets can be generated in a number of ways, all through recombinant engineering [Lee et al. 2010]. Human antigens can be injected into other species. The production of monoclonal antibodies occurs when an animal is immunized with antigen, which stimulates B cells to make a specific antibody against the introduced antigen. Subsequently, B-lymphocytes can be harvested from lymphoid organs. These cells can then be fused with cell lines from a nonsecretory myeloma line, generating transformed cells termed hybridomas. Hybridomas secreting mAb to the antigen of interest can then be selected. Given that mAbs are foreign proteins they would normally be recognized as such by human subjects, resulting in immune recognition and anti-mAb production, which in turn could impair mAb therapeutic efficacy and lead to adverse effects. With genetic engineering technology, mAbs can be modified (humanized) to decrease the content of foreign material and thereby decrease their potential immunogenicity [Weiner et al. 2010].
Chimeric mAb consist of Fab regions from another species combined with the Fc portion of a human antibody, resulting in a construct that is approximately two thirds human. Humanized antibodies consist of only the CDRs from the foreign mAb, and are about 95% human. Human mAb, where the sequence is entirely human, can be generated in a number of ways including immunization of transgenic animals that have a human immune system, repertoire cloning, and other techniques.
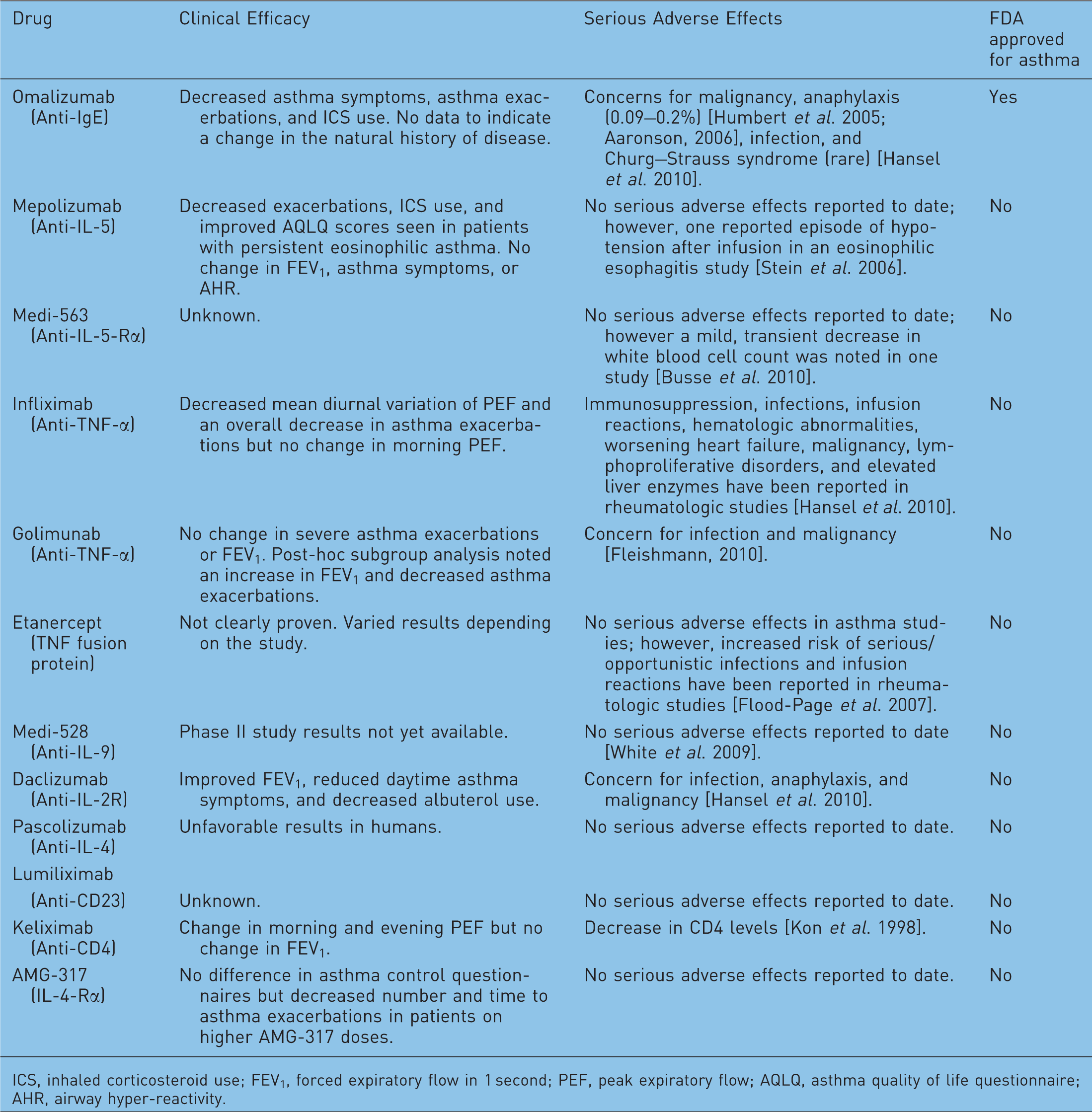
Monoclonal antibodies have been studied as potential therapeutic agents for many disease states, including allergic diseases and asthma. For asthma, humanized monoclonal antibodies that are targeted to specific mediators such as IgE, interleukin (IL)-5, IL-4, and tumor necrosis factor (TNF)-alpha have been developed and investigated [Martin-Mateos, 2007]. One of the significant limitations in studying mAbs for the treatment of severe asthma is the heterogeneity of asthma as a disease state. This issue leads to the absolute necessity for careful patient selection. There is also a need for phenotyping of patients in order to help predict potential responders to any given therapy, including mAbs. This could potentially be done through the use of biomarkers, biopsy, or sputum specimens; however, more investigation is required to identify particular patient subpopulations who may benefit from any given therapy. Here, we review the role of humanized monoclonal antibodies in the treatment of severe asthma (Figure 1). It should be noted that many of the studies included in this review used different measures of clinical efficacy. As a result, each study should be examined individually as benefit in one area of efficacy does not infer benefit in others (Table 1).
Schematic of commonly studied monoclonal antibodies in asthma. IL, interleukin; MC, mast cell; TNF, tumor necrosis factor. Summary of monoclonal antibodies studied for treatment of severe asthma. ICS, inhaled corticosteroid use; FEV1, forced expiratory flow in 1 second; PEF, peak expiratory flow; AQLQ, asthma quality of life questionnaire; AHR, airway hyper-reactivity.
Omalizumab
Omalizumab is a humanized mAb that binds to the C3 epsilon domain of free IgE, thereby inhibiting its interaction with the high-affinity portion of the IgE receptor, FcεR1 [Martin-Mateos, 2007]. This blockade prevents IgE binding to mast cells and basophils, thereby inhibiting the release of mediators when antigen is encountered. In 2000, Chang proposed that anti-IgE antibodies be used for isotype-specific control of IgE in human disease [Chang, 2000]. Several studies followed including an investigation by Lin and colleagues, which demonstrated a 96% decrease in mean serum IgE after 72 hours of anti-IgE antibody therapy [Lin et al. 2004]. Another series found that sustained omalizumab therapy resulted in downregulation of the FcεR1 expression on several cell types after 4–6 months of therapy [Lin et al. 2004; Prussin et al. 2003]. Omalizumab is currently the most well-studied mAb for the treatment of asthma. As such, it was approved by the FDA in 2003 for treatment of moderate-to-severe persistent allergic asthma in patients over 12 years of age. There is a paucity of data regarding use of omalizumab for nonallergic asthma.
According to the manufacturer [Genentech, 2003], peak omalizumab serum concentrations are reached within 7–8 days of receiving subcutaneous injection of the agent. Interval dosing studies of omalizumab found a terminal half-life of 19–22 days [Hochhaus et al. 2003] with a clinical benefit noted when free IgE levels were reduced to 50 ng/ml or less. No additional benefit was appreciated when levels were lowered to less than 12 ng/ml [Hochhaus et al. 2003]. As a result it was determined by Casale and colleagues that omaluzimab must be given in molar excess of 15:1 to 20:1 relative to total baseline IgE in order to obtain adequate reduction in IgE [Casale et al. 1997].
In terms of clinical efficacy, an observational study, comprising 280 asthmatics receiving omalizumab at a median dose of 450 mg every 4 weeks, found a reduction in daily symptoms (by 76%), nocturnal symptoms (by 84%), exacerbations (by 82%), and hospitalizations (by 78%) after 6 months on therapy [Korn et al. 2009]. Previously this same group found that omalizumab, when used as an add-on therapy to inhaled or oral steroids in stable asthmatics, resulted in a significant reduction in the risk of asthma exacerbations in the severe asthma cohort with less severe exacerbations and decreased duration of exacerbations [Busse et al. 2001]. In addition, subjects on omalizumab were more likely to decrease the dose of inhaled steroid therapy (often able to decrease the dose by 50% or less or cease inhaled steroids completely) [Busse et al. 2001; Soler et al. 2001; Milgrom et al. 1999]. In concordance with these results, Humbert and colleagues found that subjects with severe asthma who received omalizumab had a 26% decrease in asthma exacerbation rate, a 50% decrease in severe exacerbations and emergency department visits, and decreased systemic corticosteroid use [Humbert et al. 2005].
Omalizumab’s ability to decrease free IgE levels to less than 10% of pretreatment levels depends on the dose, the subject’s weight, and the subject’s baseline IgE [Boulet et al. 1997]. The recommended dose is 0.016 mg/kg per international unit (IU) of IgE given every 4 weeks, at either 2-week or 4-week intervals. Currently there is no recommended dose nor adequate clinical experience for patients with a body weight greater than 150 kg or patients with a total IgE greater than or equal to 700 IU/ml [Singh and Kraft, 2008].
The total duration of treatment with anti-IgE remains a topic of debate. Bousquet and colleagues found that of a group of patients who responded appropriately to 16 weeks of omalizumab treatment, only 61% had responded after 4 weeks of therapy; however, 87% did so at 12 weeks [Bousquet et al. 2005]. As a result, a minimum duration of 12 weeks is currently recommended before determining the degree of omalizumab’s therapeutic response [Singh and Kraft, 2008]. Currently, there is no recommendation regarding the total duration of therapy. Interestingly, after omalizumab cessation, the levels of free IgE, basophil FcεRI expression, and allergen-induced histamine release return to pretreatment levels within approximately 2–10 months [Saini et al. 1999] often with recrudescence of disease. Therefore, some studies recommend long-term or life-long treatment but this remains an active and controversial debate.
Overall, the most common adverse reaction reported with omalizumab is injection site pain. However, higher incidence of malignancies (0.5% vs. 0.2%) [Genentech, 2008], increased risk for infection versus placebo (odds ratio [OR] 1.96; 95% confidence interval[C]I 0.88–4.36) [Cruz et al. 2007], increased risk of anaphylaxis (0.09–0.2%) [Limb et al. 2007; Aaronson, 2006], and rare reports of Churg–Strauss [Hansel et al. 2010] have been reported with omalizumab use. This information, along with other studies, prompted the FDA to place a black box warning for anaphylaxis in the omalizumab package insert requiring providers to specifically inform patients of the risk associated with omalizumab use.
As a result of safety issues, a multicenter study entitled ‘Evaluating the Clinical Effectiveness and Long-term Safety in Patients with Moderate to Severe Asthma’ (EXCELS) is currently underway to evaluate the long-term safety of omalizumab in patients with moderate to severe asthma [ClinicalTrials.gov: NCT00252l35]. Of note, many of the reported omalizumab related incidents of anaphylaxis were delayed more than 2 hours in onset [Limb et al. 2007]. Therefore, an Omalizumab Joint Task Force (OJTF), including the American Academy of Allergy, Asthma, and Immunology and the American College of Allergy, Asthma, and Immunology, reviewed data and recommends an observation period of 2 hours for the first three omalizumab injections and 30 minutes for injections thereafter.
At this time, there is a relative absence of predictors of omalizumab response and an absence of validated criteria to determine objective response to this therapy. In addition, there are no data to indicate that omalizumab interferes with the natural course of severe asthma as a disease state.
Mepolizumab
Mepolizumab is an anti-IL-5 high-affinity, humanized IgG1 mAb that exerts its action by blocking IL-5 binding to the alpha-chain receptor on eosinophils. IL-5 is a major cytokine integral to eosinophilic differentiation, maturation, migration, activation, and survival. IL-5 has been found in high levels in the bronchoalveolar lavage (BAL) fluid of asthmatics [Cox, 2009]. Several studies employing animal models have shown that blockade of IL-5 results in inhibition of eosinophilic inflammation and decreased airway hyperresponsiveness [Singh and Kraft, 2008]. Consequently, mepolizumab was thought to provide an innovative biologic therapy for patients with severe asthma.
The first study of mepolizumab, performed in 2000, did not find clinical efficacy but was criticized for choice of endpoint, duration of therapy, validity of study subject selection, and inadequate power [Leckie et al. 2000]. Subsequently, a placebo-controlled study conducted by Flood-Page and colleagues in 2003 investigated the use of intravenous mepolizumab, given in three infusions over the course of 8 weeks, in 24 mild asthmatics. They found that mepolizumab eliminated eosinophils in the circulation and reduced eosinophils in the airway and bone marrow, yet did not show any improvement in clinical measurements (airway hyperresponsiveness, forced expiratory volume in 1 second [FEV1], or peak expiratory flow [PEF]) of asthma [Flood-Page et al. 2003]. Anti-IL-5 therapy did, however, reduce deposition of extracellular membrane proteins in the bronchial subepithelial basement membrane of mild allergic asthma subjects [Flood-Page et al. 2003]. Others have shown that although blood and BAL eosinophils are depleted by an 8–12 week course of anti-IL5 therapy, tissue eosinophilia persists at approximately half of pretreatment levels.
Similarly, Kips and colleagues studied the anti-IL5 mAb, SCH55700, in subjects with severe asthma whose asthma was not controlled by chronic inhaled corticosteroids [Kips et al. 2003]. In this study, it was reported that circulating eosinophils were dramatically decreased but there was not a significant improvement in pulmonary function or in asthma symptomatology [Kips et al. 2003]. A double-blind, placebo-controlled study of 362 subjects with uncontrolled asthma did not show a statistically significant difference when subjects received three monthly intravenous infusions of either placebo, 250 mg of mepolizumab, or 750 mg of mepolizumab for a total of 12 weeks [Flood-Page et al. 2007]. Adverse effects were minimal and similar between all three study groups. There appeared to be a trend toward decreased number of exacerbations in the study group on the highest dose of mepolizumab (p = 0.065).
In a randomized, double-blind, placebo-controlled study, Haldar and colleagues studied 61 subjects with refractory eosinophilic asthma where study subjects received placebo or mepolizumab monthly for 12 months to deal with concerns that prior studies were of insufficient duration to fully assess the efficacy of this class of agent. The mepolizumab cohort had significantly fewer severe exacerbations (2 in active group vs. 3.4 in placebo; p = 0.02), a significant improvement in AQLQ scores (p = 0.02), and significantly decreased serum (p < 0.001) and sputum eosinophils (p = 0.005) [Haldar et al. 2009]. Significant change in symptoms, FEV1, or airway hyperreactivity did not occur. Another double-blind, placebo-controlled study of 20 patients with persistent sputum eosinophilia and persistent asthma despite oral corticosteroid therapy was performed by Nair and colleagues. Study subjects received mepolizumab 750 mg monthly for 5 months or placebo. The mepolizumab group had decreased exacerbations (one in the active group versus 12 in placebo), reduction in oral steroid use (active group sustained reduction of 83.4% vs. 47.7% in placebo; p = 0.04), and significantly lower sputum (p = 0.005) and blood eosinophils (p = 0.004). These improvements persisted for 8 weeks after mepolizumab was stopped [Nair et al. 2009].
The latter two studies suggest that phenotypic selection of patients and duration of treatment are crucial to evaluating a targeted therapy such as anti-IL-5, which is designed specifically to act upon eosinophils. Gruenberg and Busse suggest that patients with persistent eosinophilia despite steroid therapy may have eosinophils that are resistant to corticosteroids. Therefore, this particular group of asthma patients may benefit from an agent targeted to decrease eosinophils more than another cohort of patients [Gruenberg and Busse, 2010].
Reslizumab and SCH55700 are also anti-IL5 mAb; these will not be discussed here as they have not been used in the treatment of asthma.
Anti-TNF-alpha therapies
TNF-alpha is a key proinflammatory cytokine. Inhibition of TNF has proven to be highly effective in a variety of autoimmune disorders, especially rheumatoid arthritis and other rheumatic variants. However, studies have shown increased expression of this cytokine in those with severe asthma and associated neutrophilic inflammation [Gruenberg and Busse, 2010]. As a result, anti-TNF agents have been investigated as a potential add-on therapy for patients with severe asthma but the potential role of TNF-alpha in asthma has not been completely elucidated at this time.
Infliximab
Infliximab is a chimeric anti-TNF-alpha mAb that has been more extensively studied in pulmonary diseases other than asthma. Infliximab has been investigated for treatment of pulmonary sarcoidosis and chronic obstructive pulmonary disease (COPD). The main infliximab asthma study was a small, pilot study including 38 subjects with moderate to severe asthma on inhaled corticosteroid therapy [Erin et al. 2006]. Patients receiving 5 mg/kg of infliximab had a significant decrease in mean diurnal variation of PEF after 8 weeks (p = 0.02) and an overall decrease in exacerbations (p = 0.01); however, there was not a significant change in morning PEF, which was the primary endpoint of this study. Decreased levels of TNF-alpha (p = 0.01) were observed in the treatment group. No serious adverse effects were observed; however, larger studies are necessary to validate these results [Erin et al. 2006].
Golimumab
Golimumab is a human mAb against TNF-alpha that was evaluated in a randomized, double-blind, placebo-controlled study involving over 300 subjects with severe, uncontrolled asthma. Subjects were divided into four study groups where golimumab was administered subcutaneously at monthly doses of 50 mg, 100 mg, and 250 mg, compared to placebo. Wenzel and colleagues found no difference in severe asthma attacks or FEV1 after 24 weeks of therapy (mean ± SD for severe exacerbations, placebo: 0.5 ± 1.07 vs. combined 100 and 200 mg groups: 0.5 ± 0.97). A 12% greater increase in FEV1 and fewer exacerbations were noted in a post-hoc subgroup analysis. The active treatment group with golimumab had an increase in adverse effects including life-threatening infections, eight malignancies, and one fatality [Wenzel et al. 2009]. As a result of adverse outcomes a steering committee stopped the study at week 24, and further studies have not been performed.
Etanercept
Etanercept is not a mAb but rather a fusion protein consisting of the type II TNF receptor linked to the Fc piece of human IgG. It binds both TNF-alpha and TNF-beta [Lee et al. 2010]. Howarth and colleagues performed a 12-week uncontrolled study of 15 subjects with severe asthma who received open-label etanercept as add-on therapy to inhaled corticosteroids. They noted a statistically significant improvement in methacholine-induced airway hyperresponsiveness (provocative methacholine concentration to induce a 20% decrease in FEV1 increased from a mean of 0.21 mg/ml to 1.28 mg/ml; p = 0.033), an increase in FEV1 (baseline mean ± SE of 1.91 ± 0.5 increased to 2.16 ± 0.6 posttreatment; p = 0.01), and an improvement in quality of life scores (mean of 26 decreasing to 11 after 12 weeks of therapy; p < 0.001) [Howarth et al. 2005].
In addition, they examined TNF-alpha levels in BAL fluid, TNF-alpha immunoreactive cells by immunohistochemistry, and TNF-alpha gene expression in endobronchial biopsy specimens in normal controls, mild asthmatics, and severe asthmatics. Significantly higher concentrations of TNF-alpha were found in the severe asthma cohort with a median concentration of 160 fg/ml. Interestingly TNF levels in healthy controls (median of 117 fg/ml; p = 0.001 compared with the severe asthma group) and subjects with mild asthma (median of 111 fg/ml; p < 0.001 compared with the severe asthma group) did not differ. Increased TNF-alpha mRNA levels (severe asthmatics median of 2617 vs. 84 in the mild asthmatics; p = 0.02), and greater numbers of TNF-alpha immunoreactive cells per square millimeter on biopsy (severe asthma with median of 5.6 cells/mm2; mild asthmatics with median of 2.5 cells/mm2; p < 0.03) [Howarth et al. 2005] were noted in those with severe asthma.
A later randomized, double-blind, placebo-controlled, crossover study included subjects with refractory asthma and elevated levels of TNF-alpha. Etanercept was given weekly to 10 patients with refractory asthma [Berry et al. 2006]. The study group showed a significant increase in methacholine provocation dose (p = 0.05), a statistically significant improvement in asthma quality of life scores (by 0.85; p = 0.02), and an increase in postbronchodilator FEV1 (0.32 l increase; p = 0.01) [Berry et al. 2006]. The study also found that subjects with refractory asthma had increased expression of membrane-bound TNF-alpha, TNF-alpha receptor 1, and TNF-alpha-converting enzyme [Berry et al. 2006; Erzurum, 2006]. However, subsequent trials of this agent showed no significant benefit [Brightling et al. 2008; Morjaria et al. 2008].
Recently, Holgate and colleagues performed a randomized, double-blind, placebo-controlled, phase II study of 132 moderate-to-severe asthmatics [Holgate et al. 2010]. Subjects received 25mg of subcutaneous etanercept or placebo twice weekly for 12 weeks. Study outcomes included prebronchodilator FEV1 percentage predicted change from baseline, morning PEF, FEV1 percentage predicted, asthma control questionnaires, asthma exacerbations, and PC20 by methacholine challenge. They did not report any significant difference from placebo for any of the outcomes measured. Overall, etanercept was well tolerated; however, this drug may have a worse risk/benefit ratio in asthmatics compared with patients with rheumatoid arthritis as the benefit does not appear to outweigh medication risk. Thus, despite some promising early results it is not clear what place, if any, etanercept has in the anti-asthma clinical armamentarium.
Medi-528
Medi-528, an anti-IL-9 mAb, has been investigated in several murine studies. Mouse models of airway inflammation have shown that IL-9 is involved in stimulating bronchial mucosal mucus production and mast cell accumulation [Doherty and Broide, 2007]. This process occurs through an unknown mechanism but may be associated with epithelial cell and mast cell activation through an IL-13 inflammatory pathway. Human studies are limited. Only two phase I Medi-528 dosing studies have been undertaken in healthy, human subjects and no serious adverse events were noted. However, the results of randomized phase II studies in uncontrolled moderate to severe asthmatics are not yet available [Long, 2009a].
Daclizumab
Daclizumab is a humanized mAb that binds to the alpha subunit of CD25 (IL-2 receptor). As a result of binding to the IL-2 high-affinity receptor, the activity of IL-2 is blocked [Queen et al. 1989]. The main application for daclizumab at present is in renal transplantation. However, in vitro studies demonstrate that this antibody inhibits IL-2 antigen-dependent T-cell proliferation [Junghans et al. 1990; Depper et al. 1983], mixed lymphocyte reactions [Junghans et al. 1990; Depper et al. 1983], decreases secretion of Th1 and Th2 cytokines by activated lymphocytes [McDyer et al. 2002], blocks CD-28 dependent CD40L expression on activated T cells [McDyer et al. 2002], and antibody production by B cells [Waldmann et al. 1984]. For use in asthma, it is postulated that daclizumab may act as an anti-inflammatory agent where cytokine generation is inhibited by blocking the IL-2 receptor in activated T cells [Busse et al. 2008].
Busse and colleagues performed a randomized, double-blinded, placebo-controlled trial of daclizumab in 115 subjects with moderate to severe persistent asthma. After 12 weeks of daclizumab therapy, administered intravenously every 2 weeks, subjects receiving treatment drug had improved FEV1 (4.4% ± 1.80% in treatment group versus 1.5% ± 2.39% in the placebo group; p = 0.05), reduced daytime asthma symptoms (p = 0.018), and decreased short-acting b2-agonist use (p = 0.009) [Busse et al. 2008]. Reported side effects included an anaphylactoid reaction, varicella zoster meningitis, and breast cancer in 3 of 88 subjects receiving daclizumab in this study.
Pascolizumab
Pascolizumab is a humanized anti-IL-4 mAb that inhibits the binding of IL-4 to its receptor [Hart et al. 2002]. IL-4 is a cytokine thought to have a role in the early stages of asthma by its involvement in isotype switching to IgE production, eosinophil chemotaxis, Th2 differentiation, and effector T-cell responses [Pauwels et al. 1998; Ricci, 1994]. Inhibiting IL-4 has been implicated as a mechanism to reduce the lung inflammation and remodeling that characterize severe asthma [Hart et al. 2002].
This mAb initially showed promising results in cynomolgus monkeys, however, a phase I randomized, double-blind, placebo-controlled trial of 24 patients with mild to moderate asthma did not demonstrate clinical efficacy [Shames et al. 2001] and phase II clinical trials in human asthmatics did not show favorable results. Further development of pascolizumab was discontinued [Long, 2009b]. There were no significant adverse reactions reported.
Lumiliximab
Lumiliximab is a primatized macaque/human anti-CD23 mAb. CD23 is thought to have a possible role in the regulation of IgE production and IgE-mediated inflammatory process; as a result, investigations of this agent in asthma ensued. Lumiliximab was investigated in a single-dose, placebo-controlled phase I trial involving 24 subjects with mild to moderate persistent allergic asthma. Three to six subjects in each group received a single infusion of placebo or anti-CD23 mAb at doses of 0.5, 1, 4, 10, or 15 mg/kg. During the 12-week study, lumiliximab was well tolerated. The serum half-life of the drug increased from 2 days to 10 days with increasing doses suggesting that a weekly or monthly dosing schedule may be adequate. In addition, this study showed a 40% decrease in serum IgE levels and in vitro studies revealed reduced allergen-specific peripheral blood mononuclear cell (PBMC) proliferation and reduced IL-1β and TNF-α. However, efficacy in clinical asthma remains uncertain [Poole et al. 2005].
Keliximab
Keliximab is a chimeric mAb to CD4 that was initially thought to benefit severe asthmatics by decreasing the role of CD4 lymphocytes in the pathogenesis of chronic airway disease [Kon et al. 1998]. Kon and colleagues performed a placebo-controlled, ascending-dose study involving keliximab or placebo administered in varying doses to 22 patients. Study subjects receiving 3.0 mg/kg of study drug showed a change in morning and evening PEF (median area under the curve [AUC] of 445 vs. –82.5; p = 0.005; median AUC of 548 vs. –85; p = 0.014, respectively) but there was not a difference in FEV1. The limiting side effect was an associated reduction in CD4 count [Kon et al. 1998]. As a result of decreased CD4 levels, studies were stopped.
AMG-317
AMG-317 is an IL-4R alpha antagonist that blocks both IL-4 and IL-13 pathways. IL-4 and IL-13 are both important in allergic airway inflammation; therefore, blocking these paths through the use of a fully human mAb was thought to be advantageous as a means of asthma intervention. Thus, a randomized, double-blind, placebo-controlled, phase II study was performed by Corren and colleagues, where moderate to severe asthmatics received AMG-317 (75, 150, or 300 mg subcutaneously) or placebo weekly for 12 weeks [Corren et al. 2010]. Clinical efficacy was determined by Asthma Control Questionnaires with results expressed as mean change in score ± standard error. Placebo group mean change in score was –0.49 ± 0.09, the 75 mg treatment group was –0.43 ± 0.11, the 150 mg group was –0.58 ± 0.12, and the 300 mg group was –0.70 ± 0.09 (p = 0.25). Secondary endpoints did not show a statistically significant difference. However, decreased number and increased time to exacerbations were noted in the higher AMG-317 dose groups. AMG-317 did show clinically significant levels of activity in a subset of study subjects with more severe or uncontrolled asthma symptoms. Overall, AMG-317 was well tolerated. The conclusion of the study was that AMG-317 did not demonstrate significant clinical efficacy [Corren et al. 2010].
Other potential antibody-mediated therapies for the future
Anti-IL-5 receptor alpha antibody, Medi-563
Medi-563 is a humanized recombinant IgG1κmAb that binds to an epitope within domain 1 of the alpha-chain of the IL-5 receptor [Koike et al. 2009]. As a result of binding, it inhibits proliferation of IL-5-dependent cell lines [Kolbeck et al. 2010]. Medi-563 was developed without a fucose sugar residue in the CH2 region. This modification results in enhanced eosinophil apoptosis in vitro via antibody-dependent cell-mediated cytoxicity [Kolbeck et al. 2010].
Busse and colleagues recently performed the first multicenter, open-label, escalating dose study investigating Medi-563 in adult subjects with mild atopic asthma. Medi-563 was given intravenously at doses of 0.0003–3 mg/kg over approximately 3–30 minutes. Mean serum eosinophils decreased in a dose-dependent fashion (mean ± standard deviation eosinophil count at baseline of 0.27 ± 0.2 × 103/µl which decreased to 0.01 ± 0.0 × 103/µl 24 hours postdose). Eosinophil counts remained reduced for 8–12 weeks in subjects who received higher doses of study drug. Peripheral blood eosinophil cationic protein levels were reduced from 21.4 ± 17.2 µg/l to 10.3 ± 7.0 µg/l within 24 hours postdose [Busse et al. 2010].
A safety evaluation revealed 348 reported adverse effects across all study groups. Of those reported, 86.5% were mild and 13.5% were considered moderate in severity. No serious adverse events occurred but decreased white blood cell counts did occur in 34.1%, nasopharyngitis in 27.3%, and increased blood creatine phosphokinase in 25.0%. Mean C-reactive protein (CRP) levels increased greater than 5 fold at 24 hours post dose but returned to baseline level at the end of the study [Busse et al. 2010]. Overall the safety of Medi-563 was deemed acceptable. Further studies are planned to investigate if the decreased serum eosinophil counts will translate to clinical improvements in asthmatic patients. It will be of interest to know whether tissue eosinophils are as effectively eliminated by this drug as circulating cells.
Additional possible targets for future therapy
Possible targets for future therapy include targeting human IgE-producing B cells with apoptosis-inducing M1’-specific antibodies. This was initially discussed by Brightbill and colleagues who generated mAb against the M1’ segment of human IgE through the immunization of mice but more investigation will be required to assess this possible approach [Brightbill et al. 2010]. Additional opportunities for therapy may include anti-CD69 mAb [Miki-Hosokawa et al. 2009], targeting IL-25 [Long, 2009b], using agents that target intracellular adhesion molecules, and possibly therapies that directly act against mast cells and their mediators [Singh and Kraft, 2008].
Conclusions
At this time, omalizumab has a clearly documented role in the management of severe asthma, although its role in less severe disease remains somewhat controversial, particularly in view of the expense of this agent. However, other mAb therapies, although promising, are not ready for mainstream asthma care and further studies are required. Interestingly, in some cases, a clear biologic effect of therapy was observed, such as a reduction in eosinophil numbers, yet clinical efficacy was not achieved. It would appear to be essential to properly phenotype asthma patients so that those most likely to benefit from a particular agent will be selected. An additional consideration is that of cost, omalizumab can be greater than four times the cost of many inhaled corticosteroids. We can only speculate that other mAbs would be similarly priced if not offer an increased cost expenditure. This is of economic concern for patients and healthcare providers. In addition, we must also carefully consider the potential safety issues, which in many cases may outweigh any potential benefit. Lastly, specific criteria would need to be in place in order to introduce these therapies such as a secure diagnosis of severe asthma, adequate phenotyping of the patient, and uncontrolled disease despite compliance to standard treatment. Thus, we feel that the use of most of the targeted therapies under development do not, at this time, appear to be appropriate for the entire spectrum of patients with asthma and, in fact, it is likely that some of the promising agents that have been tested and failed in the clinic, might well be effective in properly selected subsets of patients with asthma.
Footnotes
Funding
Louanne M. Tourangeau, MD was supported by the NIH (training grant T32 AI 07469) during her fellowship training.
Conflict of interest statement
None declared.