
Abstract
Pulmonary mycoses are among the most feared infections encountered in immunocompromised patients. The problem is amplified by the increasing numbers of chronically immunocompromised patients that have substantially increased both the prevalence and clinical severity of infections caused by fungi. Moreover, fungal infections in this patient population pose challenges in diagnosis and management. Fortunately, recent advances in diagnostics and antifungal therapy, and their direct application to specific diseases, provide important new approaches to this complex and often seriously ill patient population. In this article we review the commonly occurring pulmonary fungal infections in the immunocompromised population with a particular focus on their management.
Keywords
Introduction
For the last two decades, the growing numbers and diversity of immune compromised patients including persons with HIV infection, haematological malignancy, recipients of solid organ or hematopoietic stem cell transplants or patients receiving immunosuppressive drugs for the management of autoimmune conditions have dramatically increased both the prevalence and clinical severity of invasive pulmonary fungal infections [Clark and Hajjeh, 2002; Patterson, 1999; Rinaldi, 1996]. The number of people living with HIV worldwide continued to grow in 2008, reaching an estimated 33.4 million. The total number of people living with the virus in 2008 was more than 20% higher than the number in 2000, and the prevalence was roughly threefold higher than in 1990 (AIDS epidemic updates, 2009, UNAIDS). HIV-infection severely compromises cell-mediated immunity and, as a consequence, individuals are at increased risk for a specific set of mycoses. Moreover, the increasing use of aggressive immunosuppressive treatment regimens in organ transplant recipients has resulted in profound levels of immunosuppression lasting for longer periods, thus increasing the high-risk population for invasive mycoses. There is growing evidence to suggest that critically ill patients are also immunocompromised and, therefore, at high risk of invasive mycoses [Limper, 2010; Shoham, 2010].
It is important that different groups of immunosuppressed patients are susceptible to different fungal infections. For example, the most common invasive pulmonary mycoses affecting HIV-infected patients are cryptococcosis, pneumocystis pneumonia, histoplasmosis, blastomycosis, and coccidioidomycosis [Wheat, 1995]. Aspergillosis may occur in advanced stages of HIV infection, but more commonly, causes invasive pulmonary fungal infections in patients with significant neutropenia (less than 500 neutrophils/µl for more than 10 days), solid organ or hematopoietic stem cell transplant recipients, and haematological malignancies [Silveira and Husain, 2007]. In addition, aspergillosis is the most common cause of invasive pulmonary mycoses among intensive care unit patients [Limper, 2010; Shoham, 2010] and is often fatal at the anastomotic site of a lung transplant. By contrast, although colonization with Candida spp. is common, isolated pulmonary infections are rare, occurring only in the setting of disseminated candidiasis, profound aspiration of infected material, or at an airway anastomosis, and therefore the reader is referred elsewhere for the management of disseminated candidiasis [Pappas et al. 2009]. Finally, zygomycosis are important emerging fungal lung diseases among immunocompromised individuals including patients with haematological malignancies and neutropenia, hematopoietic stem cell and solid organ transplant recipients [Kontoyiannis et al. 2010; Pappas et al. 2010; Chayakulkeeree et al. 2006].
Fortunately, the use of advanced technologies, such as advanced imaging techniques, less invasive methods for obtaining surgical biopsy, and antigen testing have increased the ability to diagnose pulmonary fungal infection, although significant challenges remain. Still, pulmonary involvement by fungal infections are among the most feared complications occurring in immunocompromised patients as they are often fatal despite antifungal therapy.
Since its discovery in 1956, the polyene antifungal agent amphotericin B deoxycholate was the standard therapy for invasive fungal infections. Unfortunately, amphotericin B deoxycholate is often poorly tolerated and associated with infusion-related acute reactions and nephrotoxicity. In the late 1980s, the introduction of effective azoles (fluconazole and itraconazole), a class of effective antifungal agents that inhibits ergosterol synthesis, provided an alternative therapeutic strategy to amphotericin B deoxycholate. In recent years, several new antifungal agents have become available offering additional therapeutic options for the management of invasive fungal infections. These include lipid formulations of amphotericin B (colloidal dispersion, lipid-complex, and liposomal formulations), new azoles (voriconazole and posaconazole) and echinocandins (caspofungin, micafungin, and anidulafungin) [Denning, 2003; Johnson and Kauffman, 2003; Walsh et al. 1999; White et al. 1998].
Aspergillosis
Aspergillosis is the most common invasive filamentous fungal (mold) infection observed in the immunocompromised population. Infection is usually acquired through inhalation of the conidia of the ubiquitous soil dwelling fungus, Aspergillus spp. There are approximately 200 recognized species of Aspergillus; fortunately, only a few are known to be pathogenic to humans. Aspergillus fumigatus is by far the most common species to cause invasive pulmonary aspergillosis, followed by A. flavus and A. terreus [Morgan et al. 2005; Denning, 1998]. Only rarely does A. niger cause invasive pulmonary aspergillosis [Person et al. 2010]. Depending on the immune status of the host and the presence of underlying lung disease, this fungus can cause a variety of clinical syndromes in the lung including invasive pulmonary aspergillosis (IPA), chronic necrotizing pulmonary aspergillosis (semi-invasive aspergillosis), allergic bronchopulmonary aspergillosis and aspergilloma. IPA is a severe and commonly fatal disease that is seen in severely immunocompromised patients, while chronic necrotizing aspergillosis occurs in those who are more mildly immunocompromised or have chronic lung disease. By contrast, allergic bronchopulmonary aspergillosis is a hypersensitivity reaction to Aspergillus antigens that mainly affects patients with asthma and is not a disease of concern in the immunocompromised. Finally, aspergilloma is a fungal ball that develops in patients with pre-existing lung cavities [Soubani and Chandrasekar, 2002]. Focusing on immunocompromised patients, we confine our discussion to IPA and chronic necrotizing aspergillosis.
Invasive pulmonary aspergillosis
Over the last two decades, the incidence of IPA has increased due to the increase in the number of immunocompromised patients [Chamilos et al. 2006; Groll et al. 1996]. Immunocompromised patients with prolonged and severe neutropenia, recipients of hematopoietic stem cell transplants or solid organ transplants and patients with haematological malignancy, chronic granulomatous disease or advanced acquired immunodeficiency syndrome are at high risk for invasive aspergillosis [Segal and Walsh, 2006]. Invasive pulmonary aspergillosis is a leading cause of mortality among recipients of hematopoietic stem cell or solid organ transplants. More recently, the importance of this disease in patients with chronic obstructive lung disease on corticosteroid therapy has been realized [Vandewoude et al. 2006]. Despite antifungal therapy, the mortality due to IPA is very high (∼80%) in hematopoietic stem cell transplant recipients [Fukuda et al. 2003]. Autopsy studies have reported that invasive aspergillosis is the most common cause of infection-related death in hematopoietic stem cell transplant recipients [Sinko et al. 2008; Khayyata et al. 2007]. In addition, the mortality in solid organ transplant recipients due to invasive aspergillosis ranges from 65% to 92% [Singh and Husain, 2009].
IPA causes a necrotizing bronchopneumonia with invasion of lung parenchyma and blood vessels leading to haemorrhage and dissemination to other organs, most commonly to the brain, and less commonly to skin, pleura, heart, liver, and other organs [Denning, 1998]. Regrettably, management of IPA is very difficult in immunocompromised patients. Early administration of aggressive antifungal therapy in patients that have strongly suspected or confirmed invasive pulmonary aspergillosis is essential, because, without adequate therapy, IPA has high potential to progress to fatal pneumonia. In addition, IPA may also be complicated by extension to contiguous intrathoracic structures including the heart and great vessels and by dissemination to the central nervous system.
Antifungal therapy for pulmonary aspergillosis in immunocompromised hosts.

Adapted from Limper et al. 2011.
It is important to remember that voriconazole has high potential for drug interactions, which may result in reduced or increased serum concentration leading to therapeutic failure or toxicity. As voriconazole is metabolized by hepatic CYP2C19, CYP2C9, and CYP3A4 isoenzymes, agents (i.e. cyclosporine, rifabutin, rifampicin, carbamazepine, etc.) that inhibit and or induce these enzymes may alter the pharmacokinetics of voriconazole and vice versa [Scott and Simpson, 2007]. Therefore, voriconazole therapy is best guided by monitoring voriconazole concentration in serum [Pascual et al. 2008]. Predose voriconazole blood levels should be measured after 5–7 days of therapy to target values of 2–6 µg/ml [Ashley et al. 2006].
Lipid formulation of amphotericin B (3–5 mg/kg/day IV) is recommended as an alternative primary therapy in some patients where voriconazole cannot be given. In immunocompromised patients, antifungal therapy should be continued throughout the period of immunosuppression and until the infection has resolved. Reduction of immunosuppression (if possible) in transplant recipients can improve efficacy of treatment. Posaconazole, itraconazole, caspofungin, or micafungin have all been proposed for use in salvage therapy [Walsh et al. 2008].
The data from in vitro studies and animal models suggest that combination antifungal therapy might offer improved outcomes in the management of invasive aspergillosis; however, there is very limited clinical data available [Steinbach, 2006]. In a retrospective study, combination therapy with amphotericin B and itraconazole showed a better outcome compared with amphotericin B monotherapy [Popp et al. 1999]. In a recent case series combination therapy with voriconazole and caspofungin was associated with improved survival rate and decreased mortality compared to voriconazole monotherapy [Marr et al. 2004]. Randomized clinical trials are necessary to determine whether combination therapy should be used as a primary treatment for invasive aspergillosis.
Chronic necrotizing pulmonary aspergillosis (semi-invasive aspergillosis)
Chronic necrotizing pulmonary aspergillosis is a subacute infection most commonly seen in patients with impaired local defence from pre-existing pulmonary disease or in patients with more mild immunosuppression than that seen in IPA, including those with diabetes mellitus, poor nutrition, low-dose corticosteroid therapy, and those with connective tissue diseases such as rheumatoid arthritis and ankylosing spondylitis. In addition, patients with an aspergilloma may develop chronic necrotizing pulmonary aspergillosis after prolonged use of corticosteroids. Chronic necrotizing pulmonary aspergillosis is an indolent, destructive process due to invasion by Aspergillus and has cavitary, necrotizing and/or fibrosing components. Evidence supports itraconazole therapy in patients with chronic necrotizing pulmonary aspergillosis because of its documented efficacy and minimal toxicity (Table 1). Voriconazole and posaconazole are also likely to be efficacious and are preferred by some consultants, although we await evidence of superiority. Relapsing infection may occur, therefore, chronic maintenance therapy with itraconazole may be considered in patients with residual scarring.
In patients who do not respond to antifungal therapy, and certainly in those with hemoptysis, pulmonary resection can be considered [Saraceno et al. 1997; Binder et al. 1982]. As an adjunct to medical therapy, surgical removal of localized lung lesions may be useful in cases where the lesions are contiguous with the great vessels or pericardium due to the risk of invasion or massive haemoptysis. Surgery may also be indicated when the pulmonary lesions are refractory to aggressive antifungal therapy particularly in persistently immunocompromised patients [Salerno et al. 1998].
Prevention
Prevention of invasive pulmonary aspergillosis is an important issue considering the high mortality that occurs despite treatment among immunocompromised patients. Based on two large, prospective randomized studies [Cornely et al. 2007; Ullmann et al. 2007], the IDSA guidelines [Walsh et al. 2008] recommend the use of posaconazole for prophylaxis against Aspergillus in patients with acute myelogenous leukemia or myelodysplastic syndrome who are undergoing induction chemotherapy and also in hematopoietic stem cell transplant recipients with graft-versus-host disease. Inhaled amphotericin B has been suggested by some consultants to prevent pulmonary aspergillosis in high-risk populations [Reichenspurner et al. 1997]. Reduction of environmental exposures in high-risk patients using a specialized air-purifying system capable of removing Aspergillus spores, such as an HEPA filtration system has been frequently used. Implementation of a HEPA filtration system in a hospital ward showed dramatic reduction in the incidence of IPA among the neutropenic patients [Oren et al. 2001].
Cryptococcosis
Cryptococcosis is one of the leading causes of mortality and morbidity in immunocompromised hosts, particularly those infected with HIV. Patients infected with HIV that have a CD4 T-cell count less than 100/µl are at high risk for cryptococcal infection. Patients with haematological malignancies and immunosuppression due to chemotherapeutic agents or monoclonal antibodies, patients receiving corticosteroids for solid organ transplantation, and patients with diabetes mellitus are also predisposed to cryptococcosis [Pappas et al. 2001; Vilchez et al. 2001]. T-cell-depleting antibodies, such as alemtuzumab, which is approved for use in chronic lymphocytic leukemia, but is finding use in other applications is a potent immunosuppressive agent. This agent causes profound and long lasting depletion of CD4+ T cells and is associated with an increase in the risk for cryptococcosis [Silveira et al. 2007]. On the other hand, corticosteroids are associated with an increased risk of cryptococcosis in non-HIV-infected hosts [Vilchez et al. 2001]. Although cyclosporine and tacrolimus have the potential to reduce the risk of cryptococcal infection compared with other immunosuppressive agents due to their direct anticryptococcal activity [Mody et al. 1989, 1988], cryptococcosis has been reported in solid organ transplant recipients that receive these agents [Blankenship et al. 2005].
The majority of cases of cryptococcosis in immunocompromised hosts are caused by the encapsulated yeast, Cryptococcus neoformans (serotype A and D). Cryptococcal infection is usually acquired via inhalation of minimally encapsulated yeast. Once inhaled into the alveoli, the high pCO2 induces capsule synthesis, a major virulence factor. Protective immunity occurs when a T helper cell type 1 (Th1) response develops. An effective immune response can eliminate the yeast or result in the formation of dormant yeast within pulmonary granuloma. By contrast, compromised cell-mediated immunity allows more severe pulmonary involvement as well as dissemination to other organs.
Cryptococcal pulmonary involvement ranges from asymptomatic colonization or infection to severe pneumonia with respiratory failure depending on the immune status of the patient. In immunocompromised patients, the onset of symptoms is usually subacute, although rapid progression of pulmonary involvement may occur [Visnegarwala et al. 1998]. There are no specific radiographic features of pulmonary cryptococcosis [Zinck et al. 2002; Cameron et al. 1991]. In HIV-positive patients, pulmonary disease is less common than meningeal disease, but the reverse is true in HIV-negative patients [Jongwutiwes et al. 2008]. Indeed, lung involvement occurs in 39% of AIDS patients with cryptococcosis [Cameron et al. 1991], but in 54% of all solid organ transplant recipients with cryptococcosis [Singh et al. 2008]. Isolated pulmonary disease in the immunocompromised host is more likely to disseminate, suggesting the need for early antifungal therapy. Cryptococcus neoformans preferentially disseminates to the central nervous system leading to fatal meningitis and meningoencephalitis. Consequently, IDSA guidelines [Perfect et al. 2010] recommend lumbar puncture in immunosuppressed patients after CT scanning to look for signs of raised intracranial pressure. Results of a recent cohort study strongly recommend CSF analysis in solid organ transplant recipients with cryptcoccosis who have late-onset disease, fungemia or a cryptococcal antigen titer more than 1:64 even in the presence of normal mental status [Osawa et al. 2010]. In immunocompromised patients, signs and symptoms of meningitis including neck stiffness, headache, fever, and altered mental status should always raise suspicion of cryptococcal infection [Saag et al. 2000; Johnston et al. 1992]. In addition to lung and CNS, C. neoformans has the capacity to infect many other organs of the body including skin, joints, eye, and prostate [Wilson et al. 2008; Pappas et al. 2001; Sheu et al. 1998].
Treatment of pulmonary cryptococcosis in immunocompromised hosts.

Prophylactic fluconazole may be required for lifelong or may be discontinued after immune reconstitution
Therapy may need to be extended if the response is incomplete
ARDS, acute respiratory distress syndrome; CNS, central nervous system.
Adapted from Limper et al. 2011.
Maintenance therapy with fluconazole (200 mg/day orally) [Saag et al. 1999; Powderly et al. 1992; Bozzette et al. 1991] should be continued in HIV-infected patients that have CD4 counts less than 100 cells/µl. Itraconazole (200 mg, twice per day) is an alternative to fluconazole. Maintenance antifungal therapy can be discontinued during antiretroviral therapy when the CD4 T-cell count is more than 100 cells/µl, an undetectable or very low HIV RNA level is achieved and sustained for at least 3 months, and the patient is stable for 1–2 years. However, maintenance therapy should be reinstituted if the CD4 cell count decreases below 100 cells/µl.
In newly diagnosed AIDS patients, it is tempting to initiate antiretroviral therapy as soon as possible. However, it is prudent to wait 2–4 weeks after the commencement of anticryptococcal treatment to avoid an immune reconstitution inflammatory syndrome (IRIS) [Kaplan et al. 2009]. IRIS is a unique clinical complication encountered in severely immunocompromised HIV-infected patients as they initiate antiretroviral therapy and begin the process of immune reconstitution, or in organ transplant recipients who withdraw or reduce immunosuppression. The reconstituting immune system responds to antigens (not necessarily live organisms) with an overwhelming inflammatory response that paradoxically makes the manifestations of infection appear to be worse [Shelburne et al. 2005b]. The syndrome consists of worsening signs and symptoms of meningitis, cavitary pneumonia, intrathoracic lymphadenopathy, worsening pulmonary infiltrates, or sterile abscess in patients receiving antiretroviral treatment or organ transplantation [Singh et al. 2005; Shelburne et al. 2005a; Jenny-Avital and Abadi, 2002; King et al. 2002]. Corticosteroids (prednisone 30–50 mg/day) may be used to treat this condition [Perfect et al. 2010], however, because of the difficulty in distinguishing IRIS from therapeutic failure, consultation with an expert in the management of mycosis is encouraged for patients suspected of having the immune reconstitution syndrome.
Cryptococcal meningoencephalitis is frequently associated with raised intracranial pressure which is highly associated with morbidity and mortality; therefore, the management of raised intracranial pressure is a critical part of the care of patients with cryptococcal meningitis [White et al. 1992; Dismukes et al. 1987; Bennett et al. 1979]. The usual practice is to reduce raised intracranial pressure by removing CSF after excluding the presence of any cerebral mass by imaging with CT or MRI.
Isolated cryptococcal lung infection in immunocompromised patients should be treated based on disease severity. Asymptomatic or mild to moderate disease can be treated with fluconazole 400 mg daily for a minimum of 6–12 months. Itraconazole 400 mg/day can be used as an alternative. Severe clinical disease, including ARDS, should be treated similar to disseminated infection (Table 2). In North America and Europe, primary prophylactic anticryptococcal therapy is not routinely recommended in HIV-infected patients due to the routine use of HAART therapy. However, prophylactic therapy may be considered in areas with limited HAART availability, high levels of antiretroviral drug resistance, and a high burden of disease [Perfect et al. 2010]. Cryptococcal antigen screening in high-risk populations was reported to be highly effective at identifying those at risk of cryptococcal meningitis and helpful for implementation of targeted pre-emptive therapy [Jarvis et al. 2009].
Pneumocystis pneumonia
Pneumocystis pneumonia is one of the most prevalent and potentially fatal fungal lung infections in HIV-infected patients. In particular, HIV patients with a CD4 cell count less than 200 cells/µl have been shown to be at higher risk for Pneumocystis pneumonia. Although, the use of highly active antiretroviral therapy decreased the incidence of Pneumocystis pneumonia in HIV-infected individuals, it is still considered as an important AIDS-defining illness. Patients with haematological malignancies, transplant recipients, and those receiving immunosuppressive drugs for autoimmune and other inflammatory disorders are also at high risk for the development of Pneumocystis pneumonia [Sepkowitz, 2002]. Pneumocystis pneumonia is caused by Pneumocystis species which was initially misclassified as a protozoan. However, in 1988, it was definitively categorized as a fungus based on genetic and biochemical analyses [Edman et al. 1988]. Pneumocystis jiroveci is the only Pneumocystis species that can cause pneumonia in human [Thomas and Limper, 2007].
Treatment of Pneumocystis pneumonia in immunocompromised patients.

Adapted from Limper et al. 2011.
Adjunct therapy with corticosteroids is recommended for the HIV-infected patients with moderate to severe Pneumocystis pneumonia with hypoxemia (partial pressure of arterial oxygen less than 70 mmHg or the alveolar–arteriolar oxygen gradient greater than 35). Prednisone is recommended to start at a dose of 40 mg twice daily for 5 days, continue at a dose of 40 mg daily for another 5 days, followed by 20 mg daily for another 11 days. In non-HIV patients with severe Pneumocystis pneumonia and hypoxemia a higher dose of prednisone (60 mg or more/day) was associated with a better outcome in one retrospective analysis [Pareja et al. 1998]. Unfortunately, there are no definitive randomized controlled trials addressing the role of adjunctive corticosteroids in Pneumocystis pneumonia in non-HIV patients. Nonetheless, many experts recommend the use of corticosteroids in non-HIV infected patients with severe pneumonia and hypoxemia using dosing regimens as recommended for HIV patients [Carmona and Limper, 2010; Limper et al. 2011].
Immunocompromised patients who are at high risk for the development of Pneumocystis pneumonia should receive prophylactic therapy that should be continued until immune reconstitution occurs. HIV infected patient should start prophylactic treatment if their CD4 cell count is less than 200 cells/µl, prophylactic therapy can be discontinued safely in patients whose CD4 cell counts increase above 200 cells/µl for at least 3 months as a result of highly active antiretroviral therapy. Prophylaxis should be reintroduced if the CD4+ count falls below 200 cells/µl. Patients with previous Pneumocystis pneumonia should receive lifelong secondary prophylaxis, unless reconstitution of the immune system occurs. Non-HIV infected individuals who are immunosuppressed after solid organ transplantation or bone marrow transplantation, those with malignancies undergoing chemotherapy, and those receiving chronic immunosuppressive medications should also receive prophylaxis against Pneumocystis pneumonia [Carmona and Limper, 2010].
The first choice for Pneumocystis prophylaxis continues to be trimethoprim–sulfamethoxazole, which may be prescribed as one double-strength (preferred) tablet daily or one single-strength tablet daily or one double strength tablet three times per week. There are several alternative agents for those who cannot tolerate the trimethoprim–sulfamethoxazole therapy, such as atovaquone, dapsone or pentamidine. Atovaquone is recommended at a dose of 750 mg twice a day or 1500 mg once daily. Dapsone is recommended at a dose of 50mg twice a day or 100 mg once daily. Aerosolized pentamidine (300 mg monthly) is very rarely used in prophylaxis regimens and is discouraged due to higher risk of prophylaxis failure and the development of other infections [Carmona and Limper, 2010; Limper et al. 2011].
Histoplasmosis
Histoplasmosis, the most common endemic mycosis reported in immunocompromised patients, is caused by the dimorphic fungus Histoplasma capsulatum. Histoplasmosis is endemic in the Ohio and Mississippi River valleys of the United States, in Latin America and in some regions of sub-Saharan Africa [Kauffman, 2009; Loulergue et al. 2007; Gascon et al. 2005]. The most important risk factor for acquiring histoplasmosis (beyond exposure in an endemic area) is the immunosuppression caused by HIV infection [McKinsey et al. 1997]. Immunocompromised patients including patients with haematological malignancies, and those treated with tumour necrosis factor (TNF) antagonists or corticosteroids are at risk for histoplasmosis [Assi et al. 2007]. While infections occur in solid organ and stem cell transplant recipients, histoplasmosis may be less common than might be expected [Cuellar-Rodriguez et al. 2009; Kauffman, 2008; Vail et al. 2002]. Unfortunately, mortality in immunosuppressed patients continues to be high despite the best available care [Kauffman, 2008].
The primary site of infection is the lung, where the microconidia of H. capsulatum enter through inhalation. Cell-mediated immunity is responsible for containing the infection; therefore, in immunocompetent hosts, development of specific cell-mediated immunity usually results in clearance of the organism. On the other hand, in severely immunocompromised hosts, inadequate cell-mediated immunity allows the development of severe pulmonary infection which is often associated with dissemination. As a consequence of impaired cell mediated immunity, H. capsulatum disseminates in 95% of AIDS patients (90% have a CD4 count below 200/µl). By contrast, localized pulmonary infection may occur in patients who are not severely immunosuppressed and who have CD4 lymphocyte counts above 300/µl.
The initial signs and symptoms of histoplasmosis are similar to other types of community-acquired pneumonia. However, progression to more extensive pneumonia with marked hypoxemia and acute respiratory distress syndrome (ARDS) can occur quickly in immunosuppressed patients. Chest radiograms may show diffuse miliary or reticulonodular infiltration. Focal areas of consolidation or nodules are seen in approximately 10% of cases. Small numbers of nodules up to 5 cm in diameter may be seen in a minority of patients [Wheat, 1995]. Dissemination commonly involves adrenal glands, skin, mucous membrane, liver, bone marrow, gastrointestinal tract, and CNS. Patients with severe disseminated infection can present with septic shock, ARDS, disseminated intravascular coagulation, and multiorgan failure [Kauffman, 2008]. Gastrointestinal involvement results in diffuse ulcerations of the mucosa and sometimes malabsorption. Other commonly encountered manifestations of dissemination includes mucous membrane ulcers, skin lesions, hepatosplenomegaly, elevated liver enzymes, highly elevated serum ferritin and pancytopenia [Kauffman, 2009]. A rare but fatal syndrome called hemophagocytic syndrome has also been reported in disseminated histoplasmosis in immunosuppressed patients [Masri et al. 2003].
Antifungal treatment for pulmonary histoplasmosis.

Adapted from Limper et al. 2011.
Patients with only mild to moderate disease may be treated with oral itraconazole 200 mg, three times daily for 3 days, followed by twice daily for at least 12 months. Itraconazole blood levels vary widely among patients [Wheat et al. 1995]; therefore, serum concentrations of itraconazole should be monitored in order to ensure adequate drug levels. The oral solution of itraconazole is often preferred over the capsule formulation because of greater bioavailability. The capsule should be taken with food and an acidic gastric environment enhances absorption (orange juice or cola). By contrast, the bioavailability of the oral solution is greater when it is taken without food [Barone et al. 1998].
If the immunosuppression cannot be reversed, lifelong suppressive therapy with oral itraconazole (200 mg/day) may be necessary in order to prevent relapse. Maintenance therapy can be safely discontinued in AIDS patients who have a successful immunologic response to antiretroviral therapy. In order to discontinue maintenance therapy, patients should have a CD4 T-cell count of more than 150 cells/µl, negative results for fungal blood cultures, urine, and serum Histoplasma antigen level less than 2 ng/ml, and should be receiving antiretroviral therapy [Goldman et al. 2004]. However, it is important to resume secondary prophylaxis if antiretroviral therapy is failing, if the CD4 T-cell count decreases below 150 cells/µl, or if patients become nonadherent to antiretroviral therapy. It is suggested that urine and serum Histoplasma antigen level be followed during, and for 12 months following therapy as a predictor of treatment failure. It follows that immunosuppressed patients receiving lifelong maintenance therapy should be monitored as a rise in antigen level may indicate relapse [Wheat et al. 2007]. The success of antifungal therapy in the treatment of progressive disseminated histoplasmosis in immunocompromised patients with either amphotericin B or itraconazole is usually high while a high mortality occurs without therapy [Johnson et al. 2002; Wheat et al. 1995; Dismukes et al. 1992; Sarosi et al. 1971; Furcolow, 1963]. In cases where amphotericin B or itraconazole therapy fails, studies have reported a response to posaconazole [Restrepo et al. 2007; Clark et al. 2005].
Pulmonary nodules (histoplasmoma) usually cause no symptoms and are detected as incidental findings on chest X-rays or CT scan. These nodules are diagnostically challenging as they mimic malignancy, and are therefore frequently removed surgically to exclude malignancy. These nodules usually do not contain any viable organism, therefore no antifungal treatment is recommended if the patient is asymptomatic and contains only one or few isolated nodules. However, antifungal treatment is necessary for symptomatic patients with multiple or diffuse nodules [Limper et al. 2011; Wheat et al. 2007].
Primary prophylaxis with itraconazole (200 mg daily) is usually recommended in HIV-infected patients with CD4 cell counts less than 150 cells/µl living in endemic areas where the incidence of histoplasmosis is more than 10 cases per 100 patient-years [McKinsey et al. 1999].
Blastomycosis
Blastomycosis is an uncommon but potentially fatal fungal disease that is caused by the thermally dimorphic fungus Blastomyces dermatitidis. Blastomycosis is endemic in many regions of North America including the south central and south-eastern states that border the Mississippi and Ohio Rivers, the Midwestern states and Canadian provinces that border the Great Lakes, and a small area of New York and Canada adjacent to the St Lawrence Seaway [McKinnell and Pappas, 2009; Dworkin et al. 2005; Crampton et al. 2002]. Blastomycosis is relatively uncommon in immunocompromised patients compared with other endemic mycoses such as cryptococcosis, histoplasmosis, and coccidioidomycosis. Cases of blastomycosis have been reported infrequently in transplant recipients, HIV-infected patients and in patients receiving cytotoxic chemotherapy [Gauthier et al. 2007; Pappas et al. 1992; Pappas et al. 1993; Recht et al. 1982]. The incidence of blastomycosis in solid organ transplant patients was only 0.14% in an endemic area during 1986–2004 [Gauthier et al. 2007]. However, the cause of this low incidence in an endemic area is not clear.
Blastomycosis is acquired through the inhalation of conidia. The clinical spectrum of blastomycosis varies from subclinical infection to acute, subacute, or chronic pneumonia and disseminated disease. When it presents in the lung, less severe presentations includes lobar pneumonia, mass lesions, single or multiple nodules, and chronic fibronodular or fibrocavitary infiltrates. Acute pulmonary blastomycosis mimics community-acquired bacterial pneumonia. On the other hand, clinical features of chronic pneumonia caused by Blastomyces dermatitidis can be difficult to distinguish from tuberculosis, other fungal infections and malignancy [Chapman et al. 2008]. A subset of individuals with acute pulmonary blastomycosis can progress to fulminant multilobar pneumonia with ARDS, especially if they are immunocompromised [Lemos et al. 2001]. Although disseminated disease is uncommon in immunocompetent hosts, as high as 44% of immunocompromised patients with blastomycosis had dissemination [Pappas et al. 1993]. Outside of the lung, the usual site of dissemination includes CNS, skin, bones, and prostate. Indeed, CNS involvement was reported in 40% of patients with AIDS [Pappas et al. 1992].
Treatment options for blastomycosis.
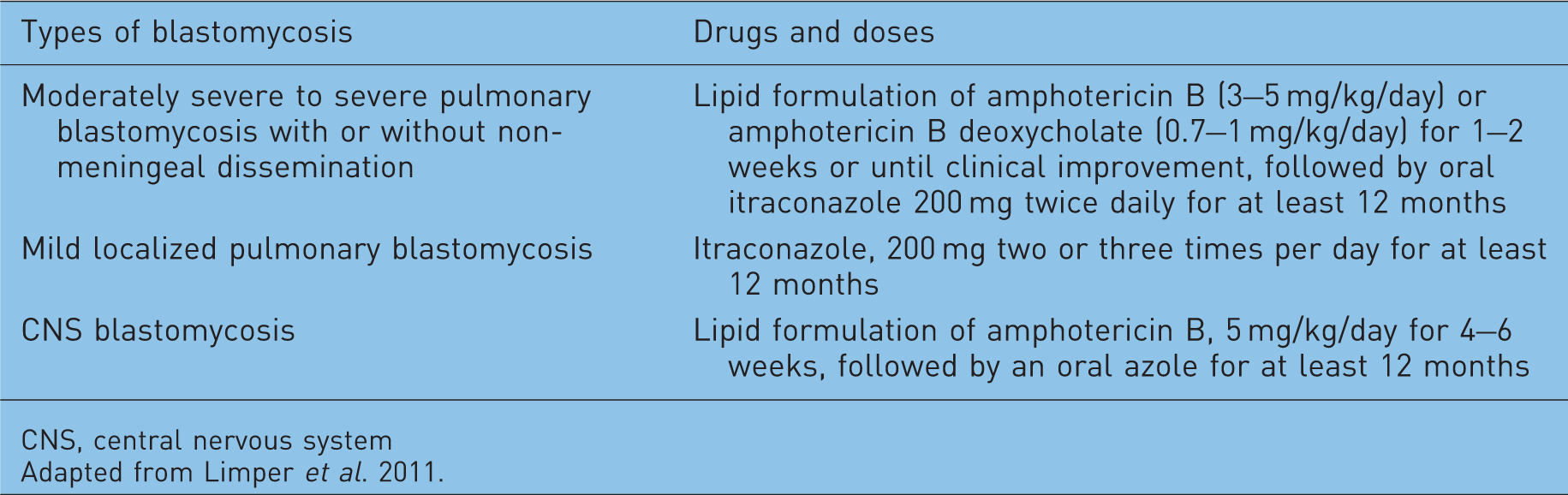
CNS, central nervous system
Adapted from Limper et al. 2011.
Coccidioidomycosis
Coccidioidomycosis (also known as valley fever) is an invasive mycosis that is endemic in certain arid and semiarid regions of the Western Hemisphere. Endemic regions for coccidioidomycosis includes the southern parts of Texas, Arizona, New Mexico, and much of Central and Southern California of the United States, semiarid areas of Mexico, and smaller endemic foci in Central and South America [Laniado-Laborin, 2007; Hector and Laniado-Laborin, 2005]. The etiologic agent of coccidioidomycosis is a dimorphic soil-dwelling fungus, Coccidioides species (C. immitis or C. posadasii) and the infection is acquired through the inhalation of the arthroconidia (spores) of this fungus. In the general population, approximately 60% of infections are asymptomatic and the vast majority of the remainder are manifested by pulmonary infection of varying severity [Smith and Beard, 1946]. The most common clinical presentation of coccidioidomycosis is a self-limiting acute or subacute community-acquired pneumonia. Approximately 5% of infections result in residual pulmonary sequelae, usually nodules or thin-walled cavities and less than 1% develop extrapulmonary diseases. The immunocompromised persons who have defective cell-mediated immunity including those with HIV infection (CD4 T-cell counts less than 250/µl), lymphoma, solid-organ transplant recipients, or recipients of rheumatologic therapies, such as high-dose corticosteroids or anti-TNF-α antibody therapy are known to be at greater risk for severe infection and dissemination. While the extrapulmonary dissemination is rare in immunocompetent individuals, 30–50% of the cases in immunocompromised patients were reported to be associated with dissemination, which is usually associated with a high rate of mortality and morbidity [Galgiani et al. 2005]. Although Coccidioides spp can disseminate to any part of the body, the most commonly affected sites includes skin, soft tissues, bones and joints, and the meninges. Meningitis is the most severe extrapulmonary complication [Parish and Blair, 2008].
Treatment options for coccidioidomycosis.

Adapted from Limper et al. 2011.
Treatment options for pulmonary zygomycosis.

Adapted from Limper et al. 2011.
Immunocompromised patients frequently present with diffuse pneumonia characterized by bilateral reticulonodular or miliary radiologic opacities. This type of pneumonia is thought to result from hematogenous spread and, therefore, requires initial aggressive antifungal therapy either with amphotericin B or high-dose fluconazole. Amphotericin B (amphotericin B dexoycholate, 0.5–1.5 mg/kg per day or alternate day, or lipid formulations of amphotericin B, 2.0–5.0 mg/kg or greater per day intravenously) is more frequently used as initial therapy if significant hypoxia is present or if deterioration is rapid. Diffuse pneumonia can progress to the ARDS, which may require mechanical ventilation. Several weeks of therapy are often required to achieve clinical improvement. Once clinical improvement is achieved, amphotericin B therapy may be discontinued and replaced with an oral azole antifungal agent. The total duration of therapy is usually at least 1 year and in case of severely immunocompromised patients, lifelong oral azole therapy should be continued in order to prevent relapse.
For the treatment of coccidioidal meningitis, fluconazole (400 mg/day) is the drug of choice currently. However, some physicians in endemic areas prefer to initiate therapy with higher doses of fluconazole (800–1000 mg daily) until clinical improvement, followed by a lower dose used as continuation therapy. Itraconazole 400–600 mg/day can be used as an alternative to fluconazole. When azole therapy fails, intrathecal amphotericin B can be used with or without concurrent azole therapy. During initial treatment, some physicians also prefer to administer intrathecal amphotericin B in addition to an azole. The latest IDSA guidelines recommend intrathecal amphotericin B, 0.1–1.5 mg/dose at intervals ranging from daily to weekly. Owing to patient intolerance, intrathecal amphotericin B should be started at a low dosage followed by a gradual increase as tolerated by the patient (limited by severe vomiting, prostration, or transient dose-related impaired status) [Galgiani et al. 2005]. Hydrocephalus is a common complication of coccidioidal meningitis and usually requires a shunt for decompression. Nonmeningeal dissemination should be treated with fluconazole or itraconazole (preferred for bone disease) or amphotericin B, and in severe cases and lifelong therapy may be required [Limper et al. 2011; Galgiani et al. 2005].
In addition to antifungal therapy, surgical debridement of large abscesses or destructive lesions may be useful [Galgiani et al. 2005]. As Coccidioidies infection frequently disseminates in immunocompromised patients and carries a high risk of mortality and morbidity, there is interest in prevention of the disease by using prophylactic antifungal therapy in high-risk populations in endemic areas. In this regard, prophylactic targeted antifungal therapy was reported to decrease the incidence of coccoidioidomycosis among solid organ transplant recipients [Vikram and Blair, 2009].
Zygomycosis
Zygomycosis has previously been considered a rare fungal infection; however, increasing numbers of invasive zygomycosis have been encountered. The growing number of immunocompromised individuals and is no longer a rare disease. The term zygomycosis describes a group of fungal infections caused by the pathogenic moulds belonging to the class Zygomycetes. The majority of the human cases of zygomycosis are caused by the Mucorale group of Zygomycetes, hence, the term ‘mucormycosis’ is frequently used to refer zygomycosis [Chayakulkeeree et al. 2006].
This fungus usually causes infections in lungs, sinuses, skin, and gastrointestinal tract. Disseminated infection may be present in severely immunocompromised patients. Pulmonary zygomycosis is most often seen in patients with hematologic malignancies and severe neutropenia, hematopoietic stem cell, and solid-organ transplant recipients. Pulmonary zygomycosis is often fatal and one study reported 76% mortality among patients with pulmonary zygomycosis in the immunocompromised population [Roden et al. 2005]. Early diagnosis and prompt administration of appropriate therapy is the key to successful management of pulmonary zygomycosis. Unfortunately, there are no randomized control trials of primary treatment of zygomycosis due to the relative rarity of the disease. Therefore, the choice of therapy has been based on experience, small case series, anecdotal case reports, information derived from animal model studies and in vitro susceptibility data. Historically, amphotericin B deoxycholate was the drug of choice, which was usually used at higher than normal doses of up to 1.5 mg/kg/day. Currently, most clinicians prefer to use lipid formulations of amphotericin B (liposomal amphotericin B and amphotericin B lipid complex) at 5 mg/kg/day as the first-line therapy due to their similar efficacy but lower toxicity. In patients who are intolerant or refractory to amphotericin B treatment, posaconazole (400 mg twice a day, orally) is an alternative (Table 7) [Limper et al. 2011]. The optimal duration of treatment is unknown but usually continues for at least 6–8 weeks [Chayakulkeeree et al. 2006].
Owing to the angioinvasive nature of these organisms, pulmonary zygomycosis often progresses to invade the major pulmonary blood vessels, adjacent organs of the chest wall, such as the mediastinum and pericardium. In addition to antifungal therapy, surgical debridement of the infected tissue should be considered urgently in these cases. In patients with pulmonary zygomycosis, surgical treatment in conjunction with antifungal therapy has been shown to improve survival significantly when compared with antifungal therapy alone [Roden et al. 2005; Lee et al. 1999].
Summary
Invasive mycoses are still a major threat to immunocompromised individuals despite recent advancements in antifungal therapy. While immunocompromised patients with prolonged and severe neutropenia are at high risk for invasive pulmonary aspergillosis, those with defects in T-cell-mediated immunity are at high risk for cryptococcosis, histoplasmosis, blastomycosis, and coccidioidomycosis. Although the availability of HAART therapy has decreased the incidence of invasive fungal infection among HIV-infected patients in North America and other developed countries, fungal infections continue to be one of the major causes of death in HIV-infected patients throughout the world. Although, the lung is the usual primary site of infection in most cases, dissemination to the CNS is the most common and serious complication in immunocompromised patients. Involvement of the central nervous system is associated with a high rate of mortality and morbidity despite therapy, therefore early diagnosis and prevention of dissemination by means of prompt antifungal therapy will help to improve the outcome of fungal diseases in immunocompromised patients. Primary prophylaxis has also been effective in reducing the incidence of invasive fungal infection among high risk patients. Development of an effective vaccine will be the next important solution to this major problem among immunocompromised patients.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors. CHM is the Jessie Boden Lloyd Professor of Immunology. AI is supported by a QEII Scholarship.
Conflict of interest statement
C.H.M. reported serving as a consultant to AstraZeneca ($1,001–$5,000) and on advisory boards for GlaxoSmithKline and AstraZeneca ($1,001–$5,000 each); he received lecture fees from AstraZeneca, Bayer, GlaxoSmithKline, Novartis, and Pfizer ($1,001–$5,000 each), and research support from AstraZeneca and Aradigm ($10,001–$50,000). A.I. has no conflicts to declare.