
Abstract
Background
Trained immunity, a form of long-term functional reprogramming of innate immune cells through epigenetic and metabolic changes, traditionally confers protection against infections. However, inappropriate activation by endogenous sterile stimuli can drive persistent maladaptive inflammation in non-communicable diseases (NCDs).
Objective
This systematic review synthesizes primary evidence for trained immunity in atherosclerosis, type 2 diabetes mellitus (T2DM), chronic kidney disease (CKD), and neurodegenerative disorders, focusing on endogenous inducers, cellular mediators, mechanisms, and translational implications.
Data Sources and Methods
Following PRISMA guidelines, we included original studies demonstrating trained immunity induced by sterile endogenous signals in the targeted diseases. Narrative synthesis was performed due to heterogeneity precluding meta-analysis.
Results
Twelve primary studies met the inclusion criteria. In atherosclerosis (n = 8 studies), oxLDL, aldosterone, Western diet lipids, and post-myocardial infarction signals induced trained immunity in monocytes or macrophages and hematopoietic progenitors via H3K4me3 enrichment, mTOR/NLRP3 activation, and glycolytic/fatty acid shifts, leading to persistent cytokine hyperproduction (TNF-α, IL-6), foam cell formation, and transmissible plaque progression. In T2DM/hyperglycemia (n = 3), high glucose levels triggered MLL-mediated epigenetic reprogramming and glycolysis-dependent “metabolic memory,” which skewed myelopoiesis and accelerated atherosclerosis despite normoglycemia. In CKD (n = 1), indoxyl sulfate induced AhR-dependent arachidonic acid pathway activation with metabolic rewiring, sustaining systemic inflammation. In neurodegeneration (n = 1), peripheral stimuli caused epigenetic reprogramming in microglia, yielding hyperresponsive or tolerized states modulating amyloid-β pathology. Convergent mechanisms (H3K4me3, glycolysis, mTOR/AhR/NLRP3) highlight trained immunity as a shared driver of chronic sterile inflammation.
Conclusions
Trained immunity emerges as a unifying maladaptive mechanism perpetuating low-grade inflammation across these diseases, bridging transient endogenous insults to sustained pathology. Targeting reprogramming pathways, such as glycolysis or epigenetic inhibitors, offers promising therapeutic strategies. Expanded human studies are needed to address preclinical dominance and data gaps, particularly in CKD and neurodegeneration, where evidence is preliminary.
Keywords
Introduction
Chronic non-communicable diseases (NCDs), including atherosclerosis, type 2 diabetes mellitus (T2DM), chronic kidney disease (CKD), and neurodegenerative disorders such as Alzheimer's disease (AD), represent leading causes of global morbidity and mortality.1,2 These conditions share a common thread of persistent low-grade inflammation driven by sterile endogenous stimuli such as oxidized lipoproteins, hyperglycemia, uremic toxins, and amyloid aggregates rather than overt pathogens.3–7 Traditionally viewed as a static first-line defense lacking memory, the innate immune system has been redefined by the discovery of trained immunity (or innate immune memory): a long-term functional reprogramming of innate immune cells (monocytes or macrophages, natural killer cells, and microglia) through epigenetic and metabolic rewiring, leading to heightened responses upon rechallenge.7,8
This paradigm shift reveals how brief exposure to endogenous danger signals can induce maladaptive hyperresponsiveness, perpetuating inflammation in sterile environments and accelerating disease progression. In contrast, immune tolerance represents the opposite phenomenon, where repeated exposure to stimuli leads to a dampened or hyporesponsive state, often serving as a protective mechanism to prevent excessive tissue damage in chronic conditions.8,9 While trained immunity is typically protective against recurrent infections by enhancing host defense, it can become destructive in NCDs by driving maladaptive, non-resolving inflammation that exacerbates pathology.5,7,10,11 For instance, in atherosclerosis, oxidized low-density lipoprotein (oxLDL) and hyperglycemia trigger myeloid bias and proinflammatory cytokine production in trained monocytes, fueling plaque instability.3,7 Similarly, in T2DM, transient hyperglycemia leaves a lasting “metabolic memory” via histone modifications and glycolytic shifts, thereby exacerbating vascular complications even after glycemic control is achieved.4,6 Emerging evidence links uremic toxins like indoxyl sulfate to trained phenotypes in CKD, promoting sustained endothelial and myeloid activation. 5 In neurodegeneration, microglial priming by amyloid-β mimics trained immunity, amplifying neuroinflammation and synaptic loss, though human data remain limited. 12 Recent syntheses further emphasize the role of trained immunity in amplifying inflammatory cascades across these diseases, with shared pathways such as mTOR-glycolysis and NLRP3-IL-1β contributing to suboptimal control of inflammation and disease progression.13,14
Despite narrative syntheses highlighting these mechanisms across subsets of diseases,5–7 no systematic review has rigorously evaluated the collective evidence for trained immunity's role, cellular mediators like monocytes, NK cells, or hematopoietic progenitors; shared pathways including mTOR-glycolysis, NLRP3-IL-1β, or therapeutic potential like modulating reprogramming to induce tolerance. With primary studies proliferating and translational implications growing, such as repurposing IL-1β inhibitors or metformin, this systematic review addresses a critical gap, synthesizing dispersed data to elucidate trained immunity as a unifying maladaptive driver in these interconnected chronic inflammatory states. The focus on these specific diseases (atherosclerosis, T2DM, CKD, and neurodegenerative disorders) was deliberate, as they exemplify major NCDs interconnected by cardiometabolic and sterile inflammatory pathways, with robust evidence of endogenous danger-associated molecular patterns (DAMPs) driving trained immunity.3–5,10 Cancer was excluded due to its emphasis on adaptive immunity, tumor-specific antigens, and immunosuppressive mechanisms, which diverge from the innate memory focus here. Respiratory conditions, such as chronic obstructive pulmonary disease (COPD), often involve mixed infectious and sterile triggers, potentially confounding the sterile inflammation paradigm central to this review.15,16 This targeted scope allows for a deeper synthesis of convergent mechanisms while highlighting gaps in less-studied areas like CKD and neurodegeneration.
Methods
This systematic review was conducted and reported in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) 2020 guidelines, 17 and the PRISMA-P protocol recommendations where applicable. The PRISMA 2020 checklist that guided this systematic review is available as Supplemental File S1.
Information sources and search strategy
Electronic databases were comprehensively searched from inception through December 29, 2025, including PubMed, Scopus, and Web of Science. Gray literature was explored via Google Scholar (limited to the first 200 results). Reference lists of included studies and key narrative reviews were hand-searched for additional citations.
The search strategy combined terms for “trained immunity” (“trained immunity”, “innate immune memory”, “epigenetic reprogramming innate”, “metabolic reprogramming monocytes”) with disease-specific terms (“atherosclerosis”, “type 2 diabetes”, “chronic kidney disease”, “Alzheimer's disease”, “neurodegeneration”, “neuroinflammation”) and relevant cellular or mechanistic terms (“monocytes”, “macrophages”, “NK cells”, “microglia”, “epigenetic”, “glycolysis”). Medical Subject Headings (MeSH) and free-text terms were used, adapted for each database. No language filters were applied initially, but non-English studies were translated if deemed relevant. The search strings used for each database are available as Supplemental File S2.
Eligibility criteria
Studies were included if they showed clear evidence of trained immunity, which means long-lasting changes in the way innate immune cells (like monocytes, macrophages, natural killer cells, microglia, or hematopoietic stem or progenitor cells) respond after being stimulated again, specifically in the context of chronic inflammatory diseases that are not caused by infections. The diseases we focused on are atherosclerosis, T2DM, CKD, and neurodegenerative disorders (mainly Alzheimer's disease or similar conditions related to brain inflammation) because they all involve inflammation caused by the body's own factors and have a significant impact worldwide, while excluding diseases like cancer (which mainly involve adaptive immunity) or respiratory diseases.
Inclusion criteria were (i) original research studies (in vitro experiments on human or animal cells, ex vivo human studies, in vivo animal models, or observational or clinical studies in humans); (ii) explicit investigation or demonstration of trained immunity mechanisms (like, increased cytokine production upon rechallenge, epigenetic marks such as H3K4me3 or H3K27ac, metabolic shifts toward glycolysis, or sustained functional changes post-initial stimulus); (iii) relevance to endogenous sterile inducers (like oxLDL, hyperglycemia, uremic toxins, amyloid-β); and (iv) publication in English. No restrictions were placed on publication date to capture the evolving field since the concept's emergence around 2011. Exclusion criteria include (1) studies focused solely on infection-driven or vaccine-induced trained immunity without linkage to chronic NCDs; (2) narrative reviews, editorials, or conference abstracts without primary data; (3) studies on adaptive immunity without innate components; and (4) non-relevant diseases, for instance, acute inflammation, cancer, or autoimmune conditions.
Study selection and data extraction
Records were imported into reference management software (Zotero) for deduplication. Screening was conducted in the Covidence systematic review software. Four independent reviewers screened titles and abstracts against eligibility criteria, followed by full-text assessment. Disagreements were resolved by consensus or arbitration by a third reviewer.
Data were extracted by four independent reviewers using a standardized, pilot-tested form.18–27 Extracted items included study characteristics (author, year, design, sample size, model: human/animal/in vitro); population or disease details; inducers of trained immunity (like oxLDL, hyperglycemia); cellular players (monocytes, NK cells); outcomes (epigenetic changes, metabolic shifts, cytokine responses, disease progression markers); and therapeutic modulation evidence (for instance, inhibitors reversing training) (Supplemental File S3). Quantitative data suitable for synthesis (fold-change in cytokine production) were prioritized. Corresponding authors were contacted for missing data where feasible.
Risk of bias assessment
Quality and risk of bias were evaluated independently by Four reviewers using appropriate tools tailored to study design: SYRCLE's RoB tool for animal studies 28 ; ROBINS-I for non-randomized human studies; and a customized checklist for in vitro or ex vivo studies based on established criteria (blinding, randomization of experiments, appropriate controls). Overall evidence certainty was graded using GRADE principles adapted for mechanistic studies (Supplemental File S4).
Data synthesis and analysis
A structured narrative synthesis was performed, organized by disease, key cellular mediators, underlying mechanisms (epigenetic and metabolic reprogramming), and emerging translational implications. This approach allowed for a comprehensive integration of the heterogeneous evidence base, highlighting patterns, consistencies, and gaps across the included studies. Key findings are summarized in a Table detailing study characteristics, inducers, cellular players, mechanisms, and functional outcomes (Table 1).
Study characteristics of the twelve included studies.

Aβ (amyloid-β), AhR (aryl hydrocarbon receptor), BM (bone marrow), CKD (chronic kidney disease), DAMPs (damage-associated molecular patterns), ESRD (end-stage renal disease), H3K4me3 (trimethylation of histone 3 at lysine 4), HIF-1α (hypoxia-inducible factor 1-alpha), HSPCs (hematopoietic stem and progenitor cells), IL-1β (interleukin-1 beta), IL-6 (interleukin-6), KMT5A (lysine methyltransferase 5A), MLL (mixed lineage leukemia), mTOR (mechanistic target of rapamycin), NLRP3 (NOD-, LRR- and pyrin domain-containing 3), oxLDL (oxidized low-density lipoprotein), Runx1 (runt-related transcription factor 1), SYK (spleen tyrosine kinase), T2DM (type 2 diabetes mellitus), TNF-α (tumor necrosis factor-alpha).
Given the marked heterogeneity in study designs (in vitro human monocyte models, ex vivo patient-derived cells, and in vivo murine disease models), experimental paradigms (varying inducers, rechallenge stimuli, and timing), and outcome measures (cytokine production, epigenetic marks, metabolic flux, and disease-specific endpoints), quantitative meta-analysis was not feasible. Instead, the narrative synthesis emphasized qualitative patterns, including the consistent induction of proinflammatory memory by diverse endogenous sterile inducers, the central role of monocytes, macrophages, and hematopoietic progenitors, and the remarkable conservation of epigenetic and metabolic hallmarks across diseases. This qualitative synthesis effectively captures the dispersed yet converging evidence, revealing trained immunity as a unifying maladaptive mechanism in chronic NCDs. By systematically mapping cellular and molecular commonalities, it addresses the fragmented nature of the current literature. It lays a solid foundation for future translational research, including the identification of shared therapeutic targets for modulating maladaptive innate immune memory.
Results
Study selection and overview
The search was conducted up to December 29, 2025. The initial search of all the databases (PubMed, Scopus, and Web of Science) yielded a total of 1036 records. After removal of 534 duplicates, the title and abstract of 502 articles were screened, leading to removal of 454 irrelevant articles. Consequently, 48 articles qualified for full-text screening, after which 39 articles were excluded that did not meet the inclusion criteria, yielding 9 eligible articles. In addition to this, 100 articles were retrieved from Google Scholar (limited to the first 200 results), out of which 78 irrelevant articles were removed during title/abstract screening, leaving 22 records eligible for full-text screening. Of these, 19 were excluded for not meeting the eligibility criteria after full-text review, yielding 3 eligible articles. In total, 12 articles were included in this systematic review. (Figure 1).

Selection process by PRISMA chart.
Characteristics of included studies
Although the search period encompassed the entire year 2025, no primary study published in 2025 met the strict inclusion criteria (original research demonstrating trained immunity induced by sterile endogenous signals in the targeted diseases). Relevant 2025 publications were primarily narrative reviews, mechanistic overviews, or studies that did not provide primary evidence of trained immunity in atherosclerosis, T2DM, CKD, or neurodegeneration, such as reviews on trained immunity modulators or DAMPs in chronic inflammation. Thus, all included studies spanned publications from 2014 to 2024, underscoring the emergence and consolidation of trained immunity as a mechanistic framework in sterile chronic inflammation. Study designs predominantly comprised in vitro and ex vivo experiments using human monocytes/macrophages, supplemented by in vivo murine models (like Ldlr⁻/⁻ mice for atherosclerosis progression and amyloid-β pathology models for neuroinflammation). One study incorporated ex vivo analyses of clinical samples from patients with end-stage renal disease. Atherosclerosis emerged as the most represented disease (8 studies), reflecting the maturity of evidence in this domain. The T2DM/hyperglycemia was addressed in 3 studies (with substantial overlap involving diabetic atherosclerosis), chronic kidney disease in 1 study, and neurodegenerative disorders (Alzheimer's disease models) in 1 study. One additional study explored maladaptive trained myelopoiesis, linking it to broader inflammatory comorbidities, including metabolic and cardiovascular conditions.
Synthesis of evidence by disease
Atherosclerosis
The majority of included studies (n = 8) presented a remarkably consistent and compelling narrative. Endogenous sterile inducers, including oxLDL, Western diet-induced hyperlipidemia, circulating aldosterone, and damage signals released after myocardial infarction, act as powerful triggers of maladaptive trained immunity, converting monocytes, macrophages, and their hematopoietic progenitors into relentless drivers of vascular inflammation. These seemingly innocuous metabolic byproducts or physiological stressors initiate a profound and durable cellular transformation through intertwined epigenetic and metabolic reprogramming. Hallmark features include enrichment of trimethylation on histone 3 lysine 4 (H3K4me3) at promoters of proinflammatory genes, mTOR-dependent oxidative stress responses, enhanced aerobic glycolysis, upregulated fatty acid synthesis, and mitochondrial metabolic alterations.30–33,38 Far from transient activation, this reprogramming establishes a stable hyperresponsive state that persists long after the initial stimulus has resolved.
The functional consequences are dramatic and clinically profound. Trained monocytes and macrophages display exaggerated production of proinflammatory cytokines such as TNF-α and IL-6 upon secondary challenge, accelerate foam cell formation, a critical step in plaque initiation, and drive markedly worsened atherosclerotic plaque progression in preclinical models. Strikingly, these effects prove transmissible: bone marrow transplantation from trained donors confers accelerated atherosclerosis in recipients, while plaque burden continues to advance even after dietary normalization or inducer withdrawal.29,34 Such findings reveal trained immunity as a self-perpetuating engine of chronic vascular inflammation, providing an elegant mechanistic explanation for the enduring cardiovascular risk conferred by even brief exposures to common risk factors like dyslipidemia, poor diet, or acute coronary events.
Collectively, this robust body of evidence elevates trained immunity from an intriguing immunological curiosity to a central orchestrator of atherosclerosis pathogenesis. By imprinting long-term proinflammatory memory at both peripheral and central (bone marrow) levels, it bridges acute metabolic insults to chronic, non-resolving disease, a paradigm shift that reframes how we understand the inexorable progression of atherosclerotic cardiovascular disease. These insights not only unify disparate risk factors under a common immunological framework but also spotlight promising therapeutic opportunities: reversing maladaptive training through targeted epigenetic modulators, metabolic inhibitors, or anti-cytokine strategies could potentially interrupt the vicious cycle and halt plaque evolution long after traditional risk factor modification.
Type 2 diabetes Mellitus (hyperglycemia)
The evidence for trained immunity in T2DM reveals hyperglycemia as a potent endogenous inducer of long-lasting “metabolic memory” in innate immune cells, offering a compelling explanation for the persistent cardiovascular risk observed even after intensive glycemic control. Three pivotal studies illuminated this process, demonstrating that brief or sustained exposure to high glucose concentrations reprograms monocytes, macrophages, and their hematopoietic precursors through glycolysis-dependent metabolic shifts and MLL-mediated epigenetic modifications.4,35,39 These alterations result in heightened proinflammatory cytokine production (like TNF-α, IL-6) upon rechallenge, a hallmark of trained immunity that endures ex vivo and long after restoration of normoglycemia.
Particularly striking were the translational implications: hyperglycemia-trained precursors, when transplanted via bone marrow into recipient mice, transmitted accelerated atherosclerotic plaque progression, directly linking diabetic “hyperglycemic memory” to macrovascular complications. 4 A third study extended these insights by implicating maladaptive trained myelopoiesis when hematopoietic progenitors skew toward proinflammatory myeloid lineages in broader inflammatory comorbidities, perpetuating systemic low-grade inflammation characteristic of T2DM. 39 Together, these findings reposition hyperglycemia not merely as a metabolic stressor but as a driver of heritable innate immune hyperresponsiveness, underscoring trained immunity as a key mechanism underlying the legacy effects of poor glycemic control and opening avenues for targeting epigenetic or metabolic nodes to mitigate diabetic vascular disease.
Chronic kidney disease
Although represented by only a single primary study in this review, the evidence for trained immunity in CKD is mechanistically profound and clinically provocative. The landmark investigation demonstrated that indoxyl sulfate, a gut-derived, protein-bound uremic toxin that accumulates progressively in CKD, induces trained immunity in monocytes and macrophages through a dual pathway involving aryl hydrocarbon receptor (AhR)-dependent arachidonic acid metabolism and concurrent epigenetic/metabolic rewiring. 36 This reprogramming manifested as sustained enhancement of proinflammatory cytokines (TNF-α, IL-6) upon secondary stimulation, observed both in vitro and ex vivo in cells from patients with end-stage renal disease. Notably, monocytes exposed to uremic sera recapitulated this trained phenotype, suggesting that chronic toxin burden in CKD fosters a self-reinforcing cycle of systemic inflammation. These findings illuminate why inflammation persists and exacerbates cardiovascular morbidity in CKD despite dialysis, positioning indoxyl sulfate-induced trained immunity as a potential central contributor to the intractable inflammatory milieu of uremia. The specificity and depth of this mechanistic insight, while based on limited primary evidence, emphasize an emerging paradigm where gut-kidney-immune axis dysregulation drives maladaptive innate memory, with profound implications for future therapeutic strategies aimed at toxin reduction or AhR pathway modulation.
Neurodegenerative disorders (neuroinflammation)
The evidence for trained immunity in neurodegenerative disorders, particularly Alzheimer's disease models, remains limited yet profoundly intriguing, with a single landmark preclinical study providing direct insight into innate immune memory mechanisms within the central nervous system. This study revealed that peripheral inflammatory stimuli mimicking systemic infections or sterile insults trigger long-term epigenetic reprogramming in microglia, the brain's resident innate immune cells, resulting in durable “trained-like” hyperresponsive or tolerized phenotypes that persist for months. 37 In Alzheimer's disease mouse models, this microglial innate immune memory exerted a bidirectional influence on disease progression: a trained hyperresponsive state exacerbated cerebral β-amyloidosis and synaptic loss, accelerating hallmark pathology, while an induced tolerized state conferred protection by alleviating plaque burden and neuroinflammation.
These findings challenge the traditional view of microglia as merely reactive responders, positioning them instead as custodians of long-term immunological memory capable of shaping neurological outcomes based on prior exposures. The epigenetic persistence mediated through histone modifications and altered transcriptional programs mirrors peripheral trained immunity paradigms but occurs in a privileged immune environment, suggesting that remote or early-life inflammatory events could predispose the brain to amplified neurodegenerative cascades later in life. Although human translational data are lacking and the available evidence is preclinical, the study's demonstration of modifiable microglial memory highlights a tantalizing therapeutic opportunity: strategically inducing tolerance to curb maladaptive neuroinflammation without compromising protective surveillance. While this represents the sole explicitly included study demonstrating trained-like reprogramming with rechallenge and epigenetic evidence in a sterile neurodegenerative context, the conceptual parallels to microglial “priming” (a heightened sensitization state observed across Alzheimer's, Parkinson's, and other tauopathies) underscore an emerging frontier warranting further primary research.
Key cellular players and convergent mechanisms
Across the spectrum of included studies, monocytes and macrophages emerged as the predominant cellular mediators of trained immunity, serving as the frontline effectors that integrate endogenous sterile signals and translate them into sustained proinflammatory responses. These circulating and tissue-resident cells consistently displayed hallmark reprogramming, enhanced cytokine production upon rechallenge, epigenetic opening of inflammatory gene loci, and metabolic polarization, making them central to the maladaptive inflammation observed in atherosclerosis, hyperglycemia, and chronic kidney disease. Hematopoietic stem and progenitor cells (HSPCs) in the bone marrow were frequently implicated as a critical upstream node, particularly in studies demonstrating transmissible training effects through bone marrow transplantation. This central reprogramming of myelopoiesis skews lineage commitment toward proinflammatory myeloid outputs, providing a compelling mechanism for how transient exposures to risk factors like oxLDL, hyperglycemia, or uremic toxins can impart lifelong inflammatory predisposition.4,29,34,39
In the brain, microglia assumed an analogous yet specialized role as the resident innate immune population capable of acquiring long-term memory-like states. The single study examining neuroinflammation revealed that peripheral stimuli induce durable epigenetic alterations in microglia, enabling either hyperresponsive (trained) or tolerized phenotypes that profoundly influence amyloid-β pathology in Alzheimer's models. 37 This finding extends the trained immunity paradigm beyond the hematopoietic compartment, suggesting that innate immune memory operates across tissue-specific contexts while preserving core functional principles.
At the molecular level, remarkable convergence emerged despite the diversity of inducers and diseases. Epigenetic modifications, particularly the trimethylation of histone 3 lysine 4 (H3K4me3) at the promoters of proinflammatory genes, constitute a nearly universal signature, facilitating transcriptional hyperactivity that persists beyond the initial stimulus. Metabolic rewiring complemented these changes, with shifts toward aerobic glycolysis (Warburg-like effect), enhanced fatty acid synthesis, or mitochondrial alterations sustaining the trained phenotype energetically. Key signaling pathways repeatedly surfaced as orchestrators: NLRP3 inflammasome activation in Western diet models, mTOR-dependent oxidative stress in oxLDL exposure, aryl hydrocarbon receptor (AhR) signaling in uremic toxin responses, and SYK-mediated histone modifications post-myocardial infarction.29,30,33,36
This striking mechanistic conservation shared cellular players, epigenetic marks, metabolic programs, and signaling nodes positions trained immunity as a unifying biological framework underlying persistent sterile inflammation across cardiometabolic, renal, and potentially neurodegenerative diseases. The preponderance of evidence in atherosclerosis and hyperglycemia reflects mechanistic maturity in these interconnected conditions, while the emerging yet mechanistically precise findings in CKD and suggestive microglial memory in neurodegeneration illuminate promising frontiers. Collectively, these insights elevate trained immunity from an intriguing observation to a central, therapeutically targetable driver of chronic disease pathogenesis, inviting strategies to reverse maladaptive reprogramming and restore inflammatory homeostasis.
Discussion
Unifying role of trained immunity in chronic Sterile inflammation
The findings from the 12 included primary studies (summarized in Figure 2) provide compelling evidence that trained immunity acts as a central maladaptive driver of persistent low-grade inflammation in NCDs, including atherosclerosis, T2DM, CKD, and neurodegenerative disorders. 11 This reprogramming enables monocytes (macrophages), hematopoietic progenitors, and tissue-resident cells like microglia to mount exaggerated proinflammatory responses to subsequent unrelated stimuli, effectively bridging transient exposure to endogenous sterile danger signals such as oxidized lipoproteins, hyperglycemia, uremic toxins, cholesterol crystals, or peripheral inflammatory cues with sustained, non-resolving disease progression. 40 Unlike classical adaptive immunity, which is antigen-specific and lymphocyte-mediated, trained immunity is broad, non-specific, and rooted in stable chromatin remodeling (like H3K4me3/H3K27ac enrichment), coupled with metabolic shifts toward aerobic glycolysis (Warburg effect) and altered mitochondrial function, sustaining enhanced cytokine production (TNF-α, IL-6, IL-1β) and effector functions long after the initial trigger. 41 This mechanism, evidenced across the reviewed primaries, explains the intractable inflammatory milieu in these conditions, where early or intermittent insults imprint durable hyperresponsiveness.
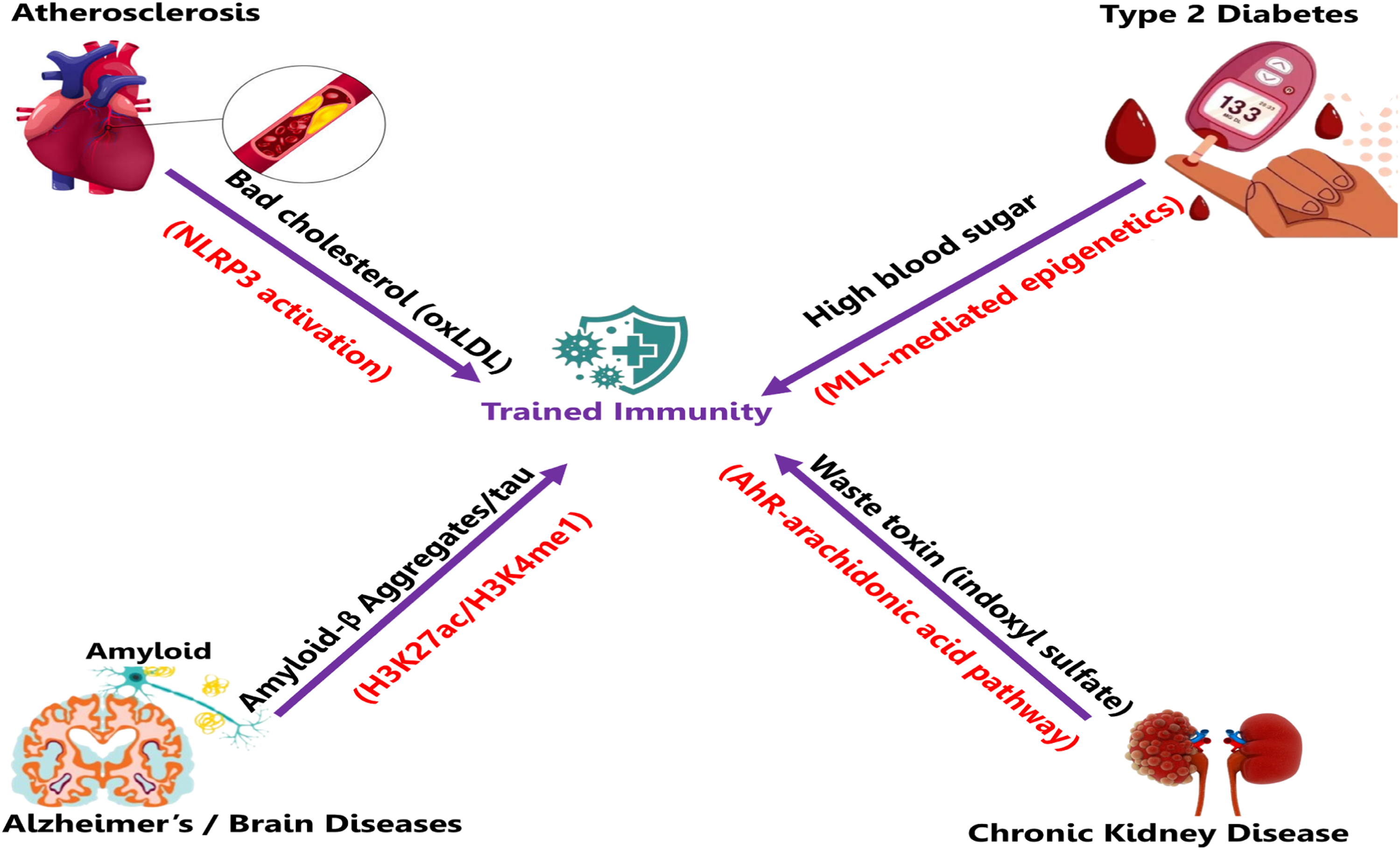
Summary of the findings.
Mechanisms in atherosclerosis: robust and convergent evidence
Atherosclerosis is supported by the largest and most consistent body of primary evidence, with eight of the twelve included studies demonstrating maladaptive trained immunity. A variety of endogenous sterile inducers, including oxLDL, lipids, and cholesterol crystals from Western diet exposure, circulating aldosterone, and damage-associated signals released after myocardial infarction, trigger durable reprogramming in circulating monocytes, tissue-resident macrophages, and bone marrow hematopoietic progenitors.29,30,32–34,38,40
The core molecular mechanisms involve H3K4me3 enrichment at promoters of proinflammatory genes, mTOR-dependent oxidative stress responses, HIF-1α stabilization, NLRP3 inflammasome activation, enhanced aerobic glycolysis, and upregulation of fatty acid synthesis pathways. 30 These changes result in functional outcomes that include exaggerated production of TNF-α and IL-6 upon secondary stimulation, accelerated formation of lipid-laden foam cells, and markedly worsened atherosclerotic plaque progression in preclinical models.29,34 A particularly striking finding is the transmissibility of the trained phenotype: bone marrow transplantation from trained donors into naïve recipients confers accelerated atherosclerosis, even in the absence of ongoing dyslipidemia or other risk factors. 40 This demonstrates that central hematopoietic reprogramming establishes a persistent vascular inflammatory risk that continues after removal of the initial inducer. Collectively, these studies unify seemingly disparate cardiovascular risk factors metabolic, hormonal, and post-ischemic, under a shared immunometabolic framework. The multi-level (peripheral and central) and multi-inducer convergence provides a mechanistic explanation for the accelerated atherosclerotic disease course observed in patients with overlapping metabolic and inflammatory comorbidities.
Mechanisms in T2DM: hyperglycemic memory and comorbidities
The evidence for trained immunity in T2DM positions hyperglycemia as a potent endogenous inducer of long-lasting “metabolic memory” in innate immune cells, providing a mechanistic explanation for the persistent cardiovascular and microvascular risk observed even after achieving glycemic control in landmark trials.4,35 Multiple studies demonstrate that brief or prolonged exposure to high glucose reprograms monocytes, macrophages, and HSPCs through glycolysis-dependent metabolic shifts characterized by the Warburg effect, increased lactate production, and HIF-1α stabilization coupled with MLL-mediated epigenetic modifications, prominently H3K4me3 enrichment at proinflammatory gene promoters.4,35
These alterations yield sustained hyperproduction of cytokines (TNF-α, IL-6, IL-1β) upon rechallenge, persisting ex vivo for days to weeks post-normoglycemia, and skew myelopoiesis toward proinflammatory myeloid lineages. 39 Bone marrow transplantation from hyperglycemic donors transmits accelerated atherosclerosis in normoglycemic recipients, directly linking this heritable innate memory to macrovascular complications. 4
Comparisons with atherosclerosis reveal a striking overlap in glycolytic/mTOR pathways, but T2DM-specific nuances, such as prominent involvement of the MLL histone methyltransferase and glutamine-dependent O-GlcNAcylation (the addition of N-acetyl glucosamine, GlcNAc) to the hydroxy group of serine/threonine residue), highlight inducer-dependent epigenetic modification.37,41 No major contradictions emerge across studies; instead, synergy reinforces trained immunity as the immunological substrate for diabetic “legacy effects,” where early hyperglycemia imprints durable hyperresponsiveness, perpetuating inflammation and complications despite later interventions. This paradigm shift opens therapeutic avenues targeting glycolysis (like metformin enhancement) or epigenetics to erase maladaptive memory and mitigate residual risk.
Emerging mechanisms in chronic kidney disease
Evidence for trained immunity in CKD remains nascent but mechanistically compelling and distinct, implicating protein-bound uremic toxins, particularly indoxyl sulfate, as endogenous inducers of durable proinflammatory memory in innate immune cells.11,36 The primary study demonstrates that indoxyl sulfate, accumulating via gut dysbiosis and impaired clearance, reprograms monocytes and macrophages through aryl hydrocarbon receptor (AhR)-dependent activation of the arachidonic acid pathway. This leads to the upregulation of arachidonate 5-lipoxygenase [ALOX5] and enhanced leukotriene signaling, with major metabolic shifts like increased glycolysis and mitochondrial respiration. 27
This results in sustained cytokine hyperproduction (TNF-α, IL-6) upon rechallenge, recapitulated in ex vivo cells from end-stage renal disease patients and uremic sera-exposed healthy monocytes, fostering a self-reinforcing inflammatory cycle. 36 Unlike glucose- or lipid-driven training in cardiometabolic diseases, the AhR-arachidonic axis represents a toxin-specific pathway, yet converges on shared metabolic endpoints (glycolytic polarization), suggesting cross-talk amplifying cardiovascular risk in CKD.11,36
Although the evidence is preliminary and derived from one core study, these findings illuminate an emerging gut-kidney-immune axis and suggest that indoxyl sulfate-induced trained immunity contributes to the intractable systemic inflammation and accelerated cardiovascular morbidity characteristic of advanced CKD. 11 Further primary research, particularly longitudinal human studies and interventional trials targeting toxin reduction or AhR pathway modulation, is essential to confirm the generalizability and therapeutic potential of these observations.
Analogous mechanisms in neurodegenerative disorders
In neurodegenerative disorders, particularly Alzheimer's disease models, evidence for innate immune memory analogous to trained immunity centers on microglial reprogramming, with a seminal preclinical study showing that peripheral or sterile inflammatory stimuli induce long-term epigenetic alterations (H3K27ac and H3K4me1 enrichment) yielding durable hyperresponsive or tolerized phenotypes persisting months. 37
These changes manifest bidirectionally: trained hyperresponsiveness exacerbates amyloid-β plaque burden, tau pathology, synaptic loss, and neuroinflammation via enhanced cytokine release and impaired clearance, while tolerance confers protection. 37 Glycolytic shifts (Warburg-like) and HIF-1α stabilization promote proinflammatory states, mirroring peripheral training but in the brain's immune-privileged environment. 42
Endogenous aggregates (amyloid-β, tau) or remote insults prime microglia, extending the paradigm centrally and suggesting early-life inflammation predisposes to amplified cascades later. 42 Though human data remain indirect and preclinical-dominant, parallels to “priming” a sensitization state with glycolytic or epigenetic hallmarks across tauopathies reinforce trained-like mechanisms perpetuating proteinopathy-neuroinflammation cycles. 12 These preliminary findings underscore the need for intensified primary research, particularly in human cohorts, to explore whether modulating microglial memory (for example, through tolerance induction) could represent a viable strategy to interrupt proteinopathy-neuroinflammation cycles.
Convergent pathways, comorbidities, and therapeutic implications
Remarkable mechanistic convergence emerges across the twelve primary studies, despite the diversity of inducers and diseases: H3K4me3 enrichment at proinflammatory gene promoters, glycolytic polarization (Warburg-like shift with HIF-1α stabilization), and orchestration by key signaling nodes such as mTOR, NLRP3 inflammasome, and AhR. 41 This shared immunometabolic circuitry explains the frequent comorbidities observed, such as accelerated atherosclerosis in T2DM (driven by overlapping glycolytic/mTOR pathways), CKD-associated cardiovascular events (via toxin-lipid cross-talk amplifying myeloid hyperresponsiveness), and potential systemic-central links in neurodegeneration.4,11,36,39
Inducer-specific nuances enrich rather than contradict the model: mTOR/oxidative stress predominates in oxLDL-driven atherosclerosis, AhR-arachidonic signaling dominates in uremic CKD, yet all converge on downstream endpoints of sustained cytokine hyperproduction and proinflammatory myeloid skewing. Treatments tested in earlier studies like blocking glycolysis with 2-deoxyglucose, inhibiting mTOR with rapamycin, using epigenetic drugs like BET or HDAC inhibitors, and Repurposed agents including metformin (a glycolysis/mTOR modulator) and IL-1β antagonists (downstream of NLRP3) already show promise in cardiometabolic clinical contexts, while tolerance induction strategies (via tolerogenic stimuli or low-dose DAMPs) offer conceptual potential for neurodegeneration.37,42 These findings suggest that trained immunity is a key area for treatment, highlighting the need for future research on immune-metabolic drugs to break harmful immune memories and restore a healthy inflammatory response.
Limitations and future directions
The current evidence base has important limitations, including heavy reliance on preclinical models (in vitro monocyte training, murine disease surrogates) that may not fully recapitulate human chronicity or compartmental specificity, methodological heterogeneity (varying inducers, rechallenge protocols, and endpoints) precluding quantitative meta-analysis, and moderate-to-high risk of bias risks arising from infrequent blinding, lack of sample size justification, and incomplete reporting in some studies. The lack of enough data in CKD (only one main study) and neurodegeneration (mostly based on animal studies with few human connections) shows that research is mostly at a preliminary stage on these diseases. Additional gaps include the in vivo duration and plasticity of trained phenotypes, the influence of microbiota, and potential germline transmission. Future directions should prioritize longitudinal human studies with epigenetic and metabolic profiling in patient cohorts, inducer-specific interventions (for instance, toxin reduction in CKD, aggregate targeting in neurodegeneration), and early-phase clinical trials of reprogramming modulators (like metformin extensions and tolerance inducers) to translate these mechanistic insights into patient benefit.
Conclusion
The synthesized primary evidence positions trained immunity as a unifying maladaptive mechanism perpetuating sterile chronic inflammation across atherosclerosis, T2DM, CKD, and neurodegenerative disorders. Convergent epigenetic (H3K4me3), metabolic (glycolysis), and signaling (mTOR/NLRP3/AhR) pathways drive comorbidity clustering and persistent residual risk, reframing our understanding of these high-burden NCDs and highlighting shared therapeutic nodes. While preclinical dominance and evidence gaps, particularly in CKD and neurodegeneration, necessitate caution, the tractability of reprogramming pathways offers substantial promise for novel immune-metabolic interventions. Expanded human validation and targeted clinical studies are now essential to harness trained immunity's potential and improve outcomes in these interconnected chronic diseases.
Footnotes
Authors contributions
JM, JH, MGK, CK, TB, and SM participated in the conceptualization, data curation, formal analysis, investigation, methodology, software, validation, visualization, and writing–original draft. JMN, AB, and AIA participated in the conceptualization, data curation, formal analysis, investigation, methodology, project administration, software, supervision, validation, visualization, writing original draft, and writing–review and editing.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.