
Abstract
Background
Acute Lung Injuries (ALI) are a severe consequence of influenza-induced cytokine storm that can cause respiratory failure and death. It has been demonstrated that Toll-like Receptor 4 (TLR4) is involved in cytokine storm and that TLR4−/− mice are protected against ALI. Therefore, TLR4 is a prime target for protection against ALI. FP12 is a known TLR4 antagonist that reduces TLR4-dependent immune activation and it is a promising lead compound for the treatment of innate immunity related pathologies.
Objectives
We present here the preclinical development of FP12 as an anti-inflammatory lead compound acting on influenza-induced ALI.
Methods
In vitro: We pre-treated THP-1 cells with FP12 (10 μM) for 0.5 h, then exposed to LPS (100 ng/ml) for 0 to 16 h. In some experiments, cells were simultaneously incubated with FP12 and LPS, or FP12 was added 30 min after LPS. Cytokine levels were measured by Western blot and ELISA assays. In vivo: WT C57BL/6J mice were infected with mouse-adapted influenza virus (PR8). Two days after infection, mice received either vehicle, FP7 (200 µg/mouse), or FP12 (200 µg/mouse) once daily (Day 2 to Day 6). Mice were monitored daily for survival for 14 days. Data were collected through histological staining, qRT-PCR, and ELISA assay.
Results
FP12 treatment inhibited both LPS- and HMGB1-induced TLR4 intracellular pathways (MyD88 and TRIF) leading to significantly reduced levels of a variety of proinflammatory cytokines including Type I interferon (IFN-β), highlighting its effectiveness in controlling proinflammatory protein production and reducing inflammation. FP12 protected mice therapeutically from influenza virus-induced lethality and reduced both cytokine gene expression and High Mobility Group Box 1 (HMGB1) levels in the lungs as well as ALI.
Conclusion
FP12 can antagonize TLR4 activation in vitro and protects mice from severe influenza infection, most likely by reducing the TLR4-dependent cytokine storm mediated by danger-associated molecular patterns (DAMPs).
Introduction
Acute Lung Injury (ALI) is a form of acute respiratory failure that is associated with hypoxia and proinflammatory cytokine and chemokine responses of which Acute Respiratory Distress Syndrome (ARDS) is the most severe form. In the case of influenza-induced disease, viral replication results in airway epithelial damage, triggering a severe inflammatory response mediated by innate immune cells, e.g., macrophages, that elicit a “cytokine storm” that may lead to ALI or ARDS. Early studies by Shirey et al.1,2 showed that TLR4-/- mice are highly refractory to mouse-adapted influenza A/PR/8/34 (PR8)-induced lethality. Since the lipid A region of Gram-negative lipopolysaccharide (LPS), the prototype TLR4 agonist, activates signaling by binding to the TLR4 co-receptor, MD-2, PR8-infected mice were treated therapeutically with Eritoran, a lipid A analog antagonist which competitively inhibits binding of LPS/lipid A to the MD-2 coreceptor. 3 When Eritoran was administered starting 2 days after PR8 infection of wild-type mice for 5 consecutive days, lung pathology, hypoxia, and cytokine and chemokine production were ameliorated, resulting in a significant increase in survival, even when Eritoran treatment was delayed as long as 6 days post-infection. 1 In addition to Eritoran, a number of other TLR4 antagonists that are structurally related or unrelated to LPS, and act either intracellularly or extracellularly, were also found to similarly protect against influenza-induced disease in mice and cotton rats. 4
In contrast to Gram-negative bacteria that express LPS, influenza does not express any TLR4 “pathogen-associated molecular patterns” (PAMPs). Therefore, it was hypothesized that a host-derived “danger-associated molecular pattern” (DAMP) was the primary TLR4 agonist in influenza-induced disease. Influenza-induced severe lung inflammation and lethality were shown to be mediated by a host DAMP, the High Mobility Group Box-1 (HMGB1), 2 a nuclear protein released from dying cells or secreted by stressed cells in any tissue. 5 HMGB1 was first identified as a key mediator of endotoxicity and sepsis.6–8 like LPS, the disulfide isoform of HMGB1 binds to MD-2 in the agonist mode and thereby activates TLR4 signaling.9,10 later studies revealed that HMGB1 is a critical mediator of endotoxicity and cecal-ligation and puncture-induced sepsis and acts by delivering HMGB1-LPS complexes to the Receptor for Advanced Glycation Endproducts (RAGE). RAGE-mediated endocytosis of HMGB1-LPS complexes causes HMGB1-mediated destabilization of acidic lysosomes and, ultimately, release into the cytoplasm of cells where LPS activates caspase-11, leading to pyroptosis. 11
Although treatment of bacterial sepsis with TLR4 antagonists may be limited to select patients, there is a large potential for development of TLR4-directed therapeutics to target sterile or microbially-induced inflammation induced by DAMPs like HMGB1. In this regard, Shirey et al. 2 reported that Eritoran inhibited both LPS- and HMGB1-induced cytokine production by macrophages in vitro. Patel et al. 12 reported that Eritoran treatment of influenza-infected cotton rats resulted in a diminished serum HMGB1 levels. Because Eritoran is difficult to synthesize 13 and is delivered intravenously, 14 we sought to develop less complex molecules that would similarly inhibit TLR4 signaling and protect against influenza-induced disease. We developed two compounds, FP7 and FP12, glucosamine derivatives with two phosphate groups and two fatty acid (FA) chains differing by two carbon atoms per chain.15,16 Both FP7 and FP12 bind to MD-2 with high affinity. Surface Plasmon Resonance (SPR) experiments and in vitro competition experiments using purified MD-2 and labelled LPS have shown that FP molecules bind MD-2 with high affinity and are able to displace LPS from MD-2. 16 FP compounds inhibit human and murine TLR4 activation by LPS in HEK-hTLR4 and RAW cells and in vivo. 16 FP7 inhibited the secretion of proinflammatory cytokines in LPS-stimulated human monocyte-derived dendritic cells with an IC50 below 1 μM.16,17 FP7 also prevented LPS-induced secretion of IFN-β, suggesting that FP7 blocked both surface MyD88-Biased TLR4 and endosomal TRIF-Biased TLR4-mediated signaling. Because FP7, like Eritoran, competes with LPS for binding to the TLR4 co-receptor, MD-2, and prevents ligand-mediated TLR4 internalization, 18 these results suggest that FP7 may not directly inhibit endosomal TLR4 TRIF-mediated signaling, but rather, inhibits upstream activation of TLR4 receptor by LPS.
We previously showed that FP7 treatment of mice after PR8 virus infection protected against lethality that is mainly due to TLR4 over-stimulation by endogenous DAMPs (e.g., HMGB-1) derived from viral damage to lung tissue. 17 Herein, we extend our studies to compound FP12, an FP7 variant, towards a drug lead targeting ALI-induced TLR4 hyper-activation and influenza-induced lethality in a murine model. FP12 presents better solubility properties than FP7, so it was selected as the best candidate to carry out the preclinical development. We investigated FP12's mechanism of action in human THP-1-derived macrophages, and assessed its efficacy in a murine model of influenza virus infection.
Materials and methods
Reagents
TLR4 antagonist FP12 was synthesized by multistep organic synthesis and the purity and identity of the compounds was confirmed by NMR, mass spectrometry and HPLC analyses (16). For TLR4-exclusive and potent activation, LPS (Salmonella Minnesota (S form), was used (Innaxon Biosciences, Tewkesbury, UK). For in vitro experiments with human and murine macrophage cell lines, FP12 was reconstituted in DMSO/ethanol (1:1) (vol: vol). For in vitro studies using primary murine macrophages, highly purified E. coli K235 lPS (prepared by Dr. Vogel) or disufide-HMGB1 (kindly provided by the laboratory of Dr. Kevin Tracey) were used as stimulants. FP12 was reconstituted in pyrogen-free saline with sonication for these studies.
Mice
All experiments using mice for the influenza studies presented herein were approved by institutional review at the University of Maryland Baltimore. Thioglycollate-elicited C57BL/6J peritoneal macrophage cultures were prepared as previously reported. 2
Cell maintenance and treatment
THP-1 cells were obtained from the European Collection of Animal Cell Cultures (Salisbury, Wiltshire, UK) and cultured in RPMI (+10% heat inactivated fetal bovine serum (HIFBS), + 1% Glutamine, + 1% penicillin/streptomycin Give final concentrations). Cells were split 3 times weekly and maintained at a density of ∼3 × 105 cells/ml. For differentiation of THP-1 cells, 25 nM of phorbol 12-myristate 13-acetate (PMA) was added to plated cells for 3 days before washing three times with fresh medium. Cells were then left to rest overnight before treatment. All cells were pre-treated with FP12 (10 μM) for 0.5 h, then exposed to LPS (100 ng/ml) for 0 to 16 h. In some experiments, cells were simultaneously incubated with FP12 and LPS, or FP12 was added to culture medium 30 min after LPS.
Mouse macrophage studies
Peritoneal macrophages isolated from C57BL/6J mice 4 days after i.p. injection of sterile 3% thioglycollate were cultured as described previously. 2 Macrophages were pretreated with medium only, FP12 (100, 1000 or 5000 ng/ml; 1000 ng/ml = 1.26 mM) for 1 h, followed by LPS (10 ng/ml) or HMGB1 (1 μg/ml) for 2 h. RNA was prepared and subjected to qRT-PCR.
Western blot analysis of protein expression and phosphorylation
Cell lysates (50 μg) were separated on 7.5% TGX gels, transferred onto polyvinyldifluoride membranes (Bio-Rad, UK) and blocked using 5% (wt/vol) skimmed milk in Tris-buffered saline (TBS)/0.1% (vol/vol) tween-20 for 1 h at room temperature. Blots were incubated overnight at 4°C with primary antibodies: phospho-p38 (4511), phospho-p65 NF-κB (3031), β-actin (12262) from Cell Signaling Technology (NEB, Herts, UK) (1:1000 dilution in TBS, 1% milk). After washing in TBS/0.1% (vol/vol) tween-20, blots were incubated with HRP-conjugated secondary antibody at room temperature for 1 h in TBS/0.1% (vol/vol) Tween-20 and 5% milk. After the final wash, immunoreactivity was visualized using the chemiluminescent substrate ECL Plus (Bio-Rad, UK). G-Box imaging system and Genesys software (Synoptics UK) were used to visualize blots and densitometry analysis was performed using Genetools (Synoptics UK). The level of cellular β-actin was used as a loading control.
ELISA
Human IL-6, IL-1β, TNFα, IFN-β and IP-10 production were measured in cell culture medium (10–100 µl) using ELISA kits (R&D Systems, USA and Ray-Biotech, USA) following the manufacturer's instructions. At the final stage, absorbance was measured at 450 nm using the Sunrise (Tecan Group LTD., Switzerland) microplate reader. Protein concentration was calculated using GraphPad Prism version 7.01. A value of p < 0.05 was considered significant.
Flow cytometry analysis
THP-1 derived macrophages were pre-treated with FP12 (10 mM) for 0.5 h and exposed to LPS (100 ng/ml) for 24–72 h. Cells were washed with PBS and trypsin/EDTA was used to detach and collect the cells. Collected cell pallets were washed with PBS and resuspended in 95 ml cell staining buffer (Bio Legend, UK). Tru-Stain blocking solution (5 μl) (Bio Legend) was added to each sample for 10 min at room temperature. Human antibody CD80-PE (305207, Bio Legend) was added after blocking in dilution 1:25 for each sample before resting the cells at 4°C for 20 min in the dark. Samples were then washed with PBS and transferred to 96 well plates and read using an Agilent NovoCyte Advanteon VBR Flow Cytometer and analysed using FlowJo software.
Statistical analysis for in vivo studies
Results were analyzed by one-way ANOVA followed by the post-hoc test (Tukey) for multiple comparisons using GraphPad Prism version 7.01. A value of P < 0.05 was considered significant.
Experiments involving influenza infection in mice
Six-week old, wild-type (WT) C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME). All animal experiments were conducted with institutional IACUC approvals from University of Maryland, Baltimore.
Viruses
Mouse-adapted H1N1 influenza A/PR/8/34 virus (“PR8”) (ATCC, Manassas, VA) was grown in the allantoic fluid of 10-day old embryonated chicken eggs as described 1 and was kindly provided by Dr. Donna Farber (Columbia University).
Virus challenge and treatment
For survival experiments, WT C57BL/6J mice were infected with mouse-adapted influenza virus, strain A/PR/8/34 (PR8; ∼7500 TCID50, i.n., 25 μl/nares), a dose of PR8 that kills ∼90% of infected mice. Stock solutions of FP7 and FP12 were made in pyrogen-free saline and sonicated until no particulates were visible. Two days after infection, mice received either vehicle (saline, i.v. or i.p.), FP7 (200 µg/mouse in 100 µl, i.v.), or FP12 (200 µg/mouse in 100 µl, i.v. or i.p.) once daily (Day 2 to Day 6). Mice were monitored daily for survival for 14 days. In some experiments, mice were infected and treated as shown in Figure 6(a) and euthanized 3 h after treatment at day 6 post-infection for analysis of gene expression or histopathology of lungs. The number of mice per treatment per experiment was based on a power analysis, with each experiment repeated at least once.
Histology and staining
Lungs were inflated and perfused and fixed with 4% PFA. Fixed sections (5 μm) of paraffin-embedded lungs were stained with hematoxylin and eosin (H&E). Four inflammatory parameters were scored independently from 0 to 4 for each section: peribronchiolitis (inflammatory cells, primarily lymphocytes, surrounding a bronchiole), perivasculitis (inflammatory cells, primarily lymphocytes, surrounding a blood vessel), alveolitis (inflammatory cells within alveolar spaces), and interstitial pneumonitis (increased thickness of alveolar walls associated with inflammatory cells. 19 Slides were randomized, read blindly, and scored for each parameter. Data is shown as a cumulation of the four parameters measured.
Quantitative real-time PCR (qrt-PCR) and qPCR
Total RNA isolation and qRT-PCR were performed as previously described.20,21 levels of mRNA for specific genes were normalized to the level of the housekeeping gene, Hprt, in the same samples and are expressed as “fold-increase” over the relative gene expression measured in mock-infected lungs. Influenza M1 gene expression was measured by quantitative PCR (qPCR) to quantify viral load. The levels of M1 are reported as direct cycle threshold [CT] . A higher CT value equates to less virus. 22
HMGB1 protein levels in lung homogenates
Protein levels of HMGB1 were measured by ELISA (IBL International (Catalog # ST51011; Toronto, Ontario, Canada) according to the manufacturer's protocols in supernatants of lung homogenates of mice infected with PR8 and treated with vehicle or FP12. The inferior lobe of the right lung was extracted and placed in viral buffer and homogenized with Omni tip homogenizer probes for 30 s. The homogenized samples were centrifuged for 10 min at 4°C at 3500 rpm. The cleared supernatant was transferred to a new tube and stored at −80°C until use.
Statistics for influenza studies
Statistical differences between two groups were determined using an unpaired, two-tailed Student's t test with significance set at p < 0.05. For comparisons between >3 groups, analysis was done by one-way ANOVA followed by a Tukey's multiple comparison post-hoc test with significance determined at p < 0.05. For survival studies, a Log-Rank (Mantel-Cox) test was used.
Results
FP12 inhibits MyD88-biased TLR4 signaling production of TLR4-dependent pro-inflammatory cytokines in THP-1-derived macrophages
To investigate the potential of FP12 as a TLR4 antagonist to block LPS-induced TLR4/MyD88-Biased signaling, ELISA and Western blot approaches were carried out to assess its effect on two levels: second messengers and production of TLR4 proinflammatory proteins in THP-1 macrophages. LPS-induced phosphorylation of p65 NF-κB and p38 MAPK were significantly reduced in a concentration-dependent manner following preincubation of cells with FP12 (Figure 1). While p38 MAPK phosphorylation was significantly reduced at 0.1 µM of FP12, p65 NF-κB was significantly downregulated by >5 µM FP12 (Figure 1).

FP12 downregulated LPS-induced p65 NF-κB (a) and p38 MAPK (b) phosphorylation in THP-1 macrophages. Cells were pre-treated with FP12 (0–10 µM) for 1 h before being exposed to LPS (100 ng/ml) for 1 h. Cell lysates were collected and analyzed by Western blot to determine levels of p65 NF-κB and p38 MAPK phosphorylation. B-actin was used as a loading control. Results are shown as mean ± SD of two to three separate treatments. Statistically significant results are indicated as ***p < 0.001 for control vs LPS and LPS vs LPS + FP12 treated cells.
We previously showed the potential of FP12 to downregulate production of a number of TLR4/MyD88-biased proinflammatory cytokines using semi-quantitative analysis of inflammation antibody array. 23 Here, we used an ELISA approach to validate array results for IL-1β and IL-6. Following overnight activation of TLR4 signaling with LPS, IL-1β production was significantly increased in treated cells as compared to the non-treated cells. ELISA results showed that 1 μM of FP12 was sufficient to produce a significant reduction in IL-1β secretion (Figure 2(a)).

FP12 downregulated LPS-induced IL-1β (a) and IL-6 (b) secretion in THP-1 macrophages. Cells were pre-treated with FP12 (0–10 µM) for 1 h prior to exposure to LPS (100 ng/ml). IL-1β and IL-6 production was measured using ELISA after 18 h in culture medium. Results are shown as mean ± SD of three separate treatments. Statistically significant results are indicated as ***p < 0.001 control vs LPS and LPS vs LPS/FP12 treated cells.
In a comparable manner, IL-6 production was significantly upregulated by LPS and a concentration-dependent reduction in IL-6 production was observed in THP-1 macrophages pre-treated with FP12 (Figure 2(b)). FP12 was efficient in downregulating IL-6 production with as little as 0.1 μM in THP-1 macrophages. These results clearly show that FP12 can significantly inhibit LPS-stimulated TLR4/MyD88-biased signaling in THP-1 macrophages.
We then investigated the effect of FP12 (0–10 μM) on THP-1 macrophage viability. MTT results demonstrated that FP12 (in the presence or absence of LPS) did not significantly affect THP-1 macrophage viability (Supp. Info., Figure 1S).
FP12 inhibits M1 polarization in THP-1 macrophages
Bearing in mind that FP12 downregulated production of TLR4-dependent proinflammatory mediators (biomarkers of M1 macrophage polarization), in the next series of experiments we further examined the potential of this antagonist to modulate CD80 expression (a well-known M1 biomarker) using a flow cytometry approach. Results clearly confirmed the ability of FP12 to completely block LPS/IFNγ-stimulated CD80 expression (24 h–72 h) in M1 THP-1 macrophages (Figure 3).

FP12-downregulated LPS/IFNγ-induced CD80 expression at 24 h (a), 48 h (b) and 72 h (c). Cells were pre-treated with FP12 (10 µM) for 0.5 h prior to exposure to LPS (100 ng/ml) and IFNγ (20 ng/ml). CD80 expression was measured using flow cytometry. Results are shown as mean ± SD of two separate treatments. Statistically significant results are indicated as ***p < 0.001 and **p < 0.01 for control vs LPS/IFNγ and for LPS/IFNγ vs. LPS/IFNγ /FP12 treated cells.
FP12 downregulates TLR4/TRIF-induced proinflammatory cytokines and type I interferon
LPS binds to MD-2 causing TLR4 dimerization and activation of MyD88-Biased signaling. This triggers a signaling cascade, resulting in release of downstream second messengers following LPS stimulation. FP12 is thought to interact with MD-2 in a similar manner to LPS and may prevent LPS binding to the TLR4 through a mechanism of competitive inhibition.15,16
To better understand the pharmacological action and interaction of FP12 timing on TLR4 signaling, THP-1 macrophages were treated at 30 min before, simultaneously with, and 30 min after LPS exposure. Following stimulation, the level of TNFα was significantly downregulated by FP12 in all timing treatments (Figure 4(a)).
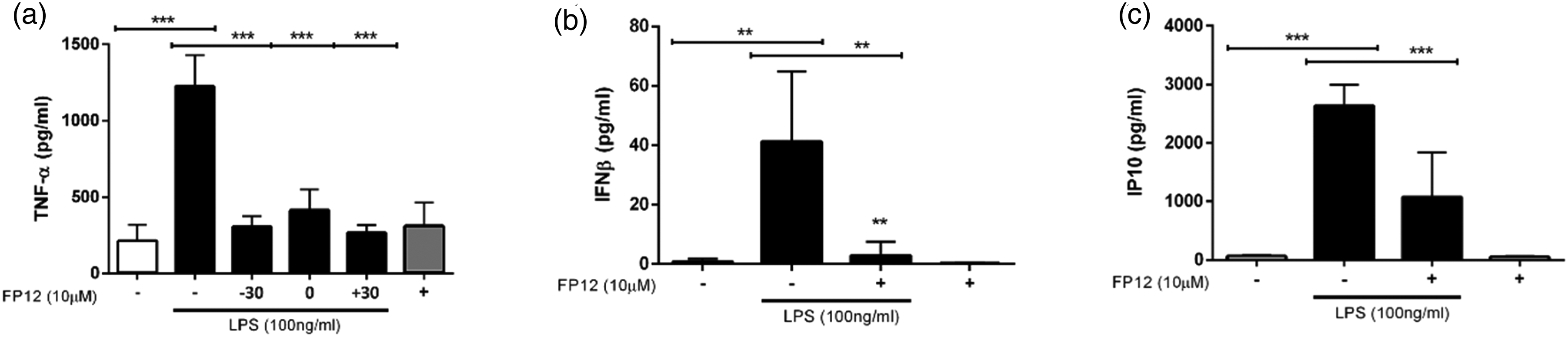
FP12 down-regulates LPS-induced MyD88- and TRIF-biased cytokines under different treatment conditions. (a) THP-1 macrophages were treated with FP12 (10 µM) 30 min prior (−30), simultaneously with (0) or 30 min after (+30) LPS (100 ng/ml) treatment of cells. (B and C). THP-1 macrophages were treated with 10 µM FP12 for 30 min before LPS (100 ng/ml) exposure at 3 h (b) and 18 h (c). Culture supernatants were collected and analyzed by ELISA. Results are shown as mean ± SD of three separate treatments. Statistically significant results are indicated as ***p < 0.001 and **p < 0.01 for control vs. LPS and LPS vs. LPS + FP12 treated samples.
TLR4-induced TRIF-biased signaling is characterized by internalization of the receptor complex 24 and downstream production of interferons including IFN-β and the chemokine IP10. In another series of experiments, we determined the effect of FP12 on LPS-stimulated IFN-β/IP-10 production using ELISA. Following exposure to LPS, IFN-β and IP-10 levels were shown to be increased significantly. Again, FP12 was capable of down-regulating LPS-induced IFN-β/IP-10 production (Figure 4(b) and (c)). Together, these results show the potential of FP12 to inhibit both MyD88- and TRIF-biased signaling in THP-1 macrophages. To confirm and extend these findings, qRT-PCR was used to analyze the effect of FP12 on LPS-induced MyD88- (Il1b, Ptgs2) and TRIF- (Ifnb, Ccl5) dependent gene expression in primary murine macrophages. FP12 also inhibits both arms of the LPS-induced signaling pathway (Supp. Info. Figure 2S.) In addition to blocking LPS-induced gene expression, FP12 also blocked cytokine gene induction (Il1b, Ptgs2, Ifnb) in murine macrophages induced by the host-derived DAMP, HMGB1, that also stimulates TLR4/MD-2 signaling (Supp. Info. Figure 3S.)
FP12 treatment blocks influenza-induced disease therapeutically in mice
Previous studies have shown that TLR4 antagonists, including FP7, blunt influenza-induced disease in mice and in cotton rats. 4 To compare the efficacy of FP12 with that of FP7, wild-type C57BL/6J mice were infected on Day 0 with a dose of mouse-adapted influenza PR8 that kills ∼90% of the animals (LD90). On Days 2–6, mice (n = 5/treatment) were treated once daily, i.v., with saline (control), FP7, or FP12 (200 μg/mouse/day) as shown in Figure 5(a). In this experiment (Figure 5(b)), both FP7 and FP12 elicited equivalent and statistically significant protection compared to saline-treated mice, with FP12 resulting in a slight delay in the meantime to death compared to FP7.
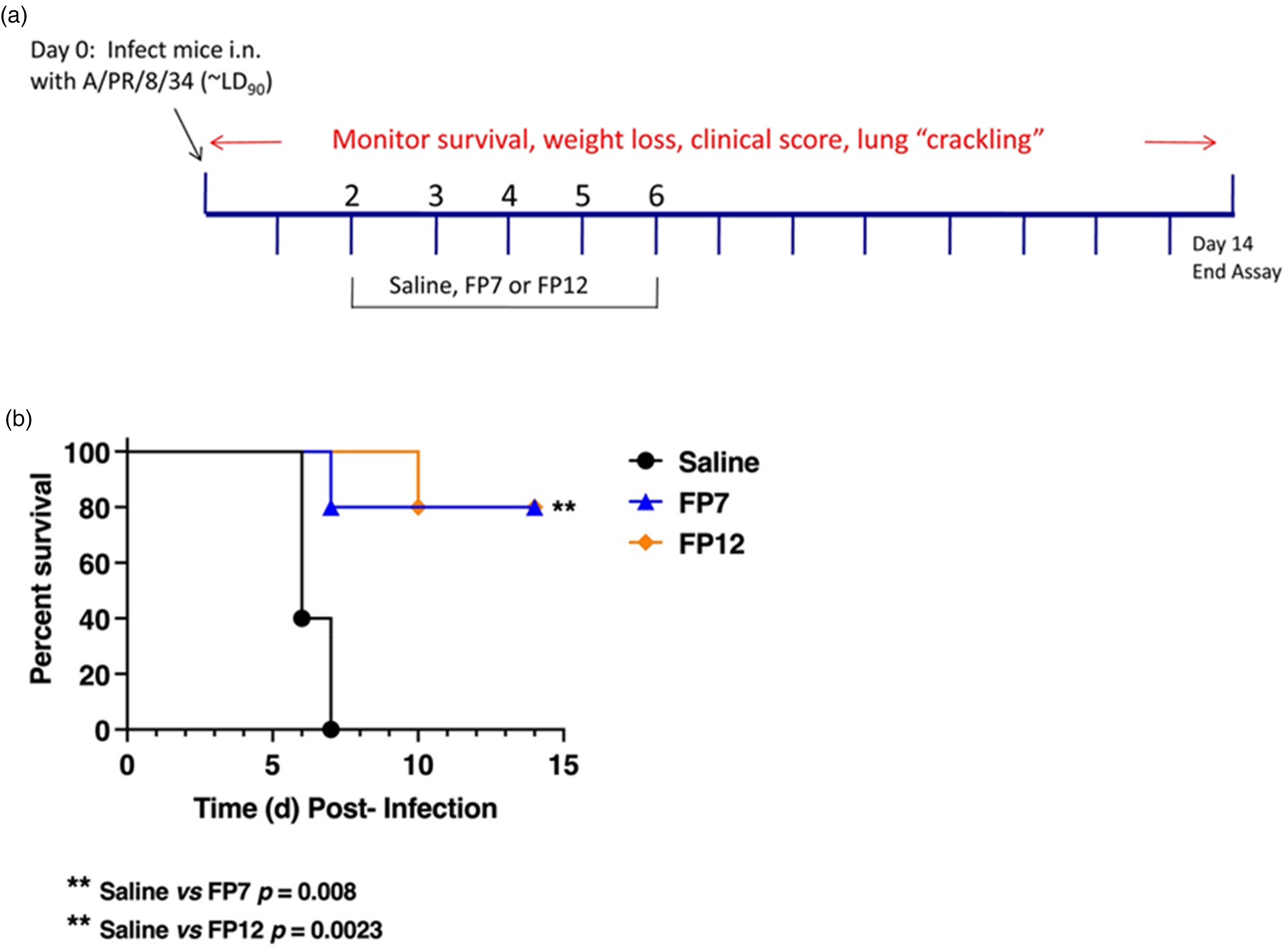
FP12 treatment protects mice from lethal influenza challenge. (a) Diagram of protocol showing infection and treatment. C57BL/6J mice were infected with mouse-adapted influenza, strain PR8 (∼7500 TCID50, i.n.; ∼LD90). Two days later, mice received vehicle (saline; i.v.), FP7 or FP12 (200 μg/mouse; i.v.) once daily from days 2 to 6 post-infection. Mice were monitored daily for survival for 14 days. (b) Survival curve of influenza-infected mice treated as described in (a) (5 mice/treatment group).
We next carried out a modification of this experiment in which the mice were treated identically as in Figure 5(b) with the exception that saline or FP12 was administered i.p. Figure 6(a) shows that the combined results of two separate experiments (n = 10 mice/treatment) recapitulate the data seen in Figure 5(b), indicating that the protection afforded by FP12 is highly significant when administered by either the i.v. or i.p. routes. The identical experiment was repeated, and the lungs of the mice were harvested at Day 6 post-infection, the day on which the control mice typically begin to die. qRT-PCR of lung cytokine mRNA, HMGB1 levels in lung homogenates, and histopathology of formalin-fixed, H&E-stained lung sections were performed (Figures 6(b)–(d)). Il1b, Il6, and Cxcl1 gene expression were significantly decreased by treatment of influenza-infected mice with FP-12, with the others (Tnf, Ifnb, and Ccl5) exhibiting the same trend (Figure 6(b)). Virus M1 mRNA levels (a relative measure of virus proliferation) were not significantly affected by FP12 treatment. Previous studies have shown that Eritoran treatment of influenza-infected mice and cotton rats resulted in decreased levels of HMGB1 in sera and lung homogenates.12,17 Treatment with FP12 significantly decreased the levels of HMGB1 (Figure 6(c)) compared with vehicle-treated mice.

Intraperitoneal administration of FP12 provides significant protection and suppresses influenza-induced acute lung injury. (a) C57BL/6J mice (10 mice per group) were infected with mouse-adapted influenza, strain PR8 (∼7500 TCID50, i.n.; ∼LD90). Two days later, mice received vehicle (saline; i.p.) or FP12 (200 μg/mouse; i.p.) once daily from days 2 to 6 post-infection. Mice were monitored daily for survival for 14 days. (5 mice/treatment group). (b and c) C57BL/6J mice were infected and treated as described in (a). On day 6 post-infection, mice were euthanized and lungs were extracted and processed for total RNA and gene expression analyzed by qRT-PCR (b), HMGB1 protein levels (c), and stained for histopathology and examined for tissue damage and inflammatory cellular infiltration (d). Representative micrographic images are shown (PR8 + vehicle on the left and PR8 + FP12 on the right). Combined mean histopathology scores for each group were determined blindly as previously described 19 (n = 5 mice/treatment group). Representative H&E-stained lung sections are shown.
Figure 6(d) shows treatment of mice with FP12 resulted in less lung pathology as shown by comparison of micrographic images. The combined histology score that was measured blindly for 4 parameters of pathology (i.e., perivasculitis, peribronchiolitis, interstitial pneumonitis, and alvelolitis), 19 was significantly reduced by treatment with FP12. Collectively, these data indicate that FP12, like Eritoran and FP7, is a potent anti-inflammatory agent and protects against an otherwise lethal influenza infection by blocking proinflammatory gene expression, HMGB1 release, and the subsequent lung inflammation. In contrast to Eritoran, however, FP12 has the added benefits that it is soluble in saline (with sonication) and can be administered therapeutically by the intraperitoneal route.
Discussion
The identification of small molecule synthetic TLR4 antagonists paves the way for inhibition of not only inflammation induced by LPS-containing Gram-negative bacteria, but also other cytokine-mediated inflammatory diseases caused by host-derived DAMPs that also signal through TLR4. The activation of TLR4 signaling by LPS is a complex process: an acute phase reactant, lipid A binding protein (LBP), binds LPS 25 and transfers LPS monomers to a non-signaling receptor, CD14, 26 where the fatty acids of the lipid A portion of LPS bind in a deep hydrophobic pocket. 3 A single
LBP molecule bound longitudinally to LPS micelles catalyzes multi-rounds of LPS transfer to CD14 s that rapidly dissociated from LPB-LPS complex upon LPS transfer via electrostatic interactions. Subsequently, the single LPS molecule bound to CD14 is transferred to TLR4-MD2 in a TLR4- dependent manner 27 The formation of TLR4/MD-2 heterodimer creates an intracellular “signaling platform” that activates two major signaling pathways, the MyD88 pathway, which occurs at the cell surface, followed by the TRIF pathway, which acts after the LPS/TLR4/MD-2/CD14 complex is endocytosed. 28 The first structurally related LPS antagonist to be synthetically produced was Eritoran (Eisai Inc.).14,29 Eritoran acts by binding deeply to the MD-2 hydrophobic pocket, thereby competitively inhibiting binding of LPS and signal transduction. 3 The synthesis of Eritoran is a multistep, highly complicated process, 13 and the final product must be administered intravenously in vivo. For these reasons, development of simpler, more soluble molecules with similar inhibitory bioactivities was undertaken.
In contrast Eritoran, which has a disaccharide backbone with four fatty acids. 29 we previously reported synthesis of FP7, a glucosamine monosaccharide with two fatty acids. 15 FP12 represents a second-generation antagonist with straightforward synthesis, improved solubility and bioactivity. In this report, we first show that FP12 inhibited LPS-induced phosphorylation of two important TLR4/MyD88 mediators, p65 NF-κB andp38 MAPK, that are required for induction of proinflammatory cytokine secretion. In addition, FP12 inhibited upregulation of the M1 biomarker, CD80, induced by LPS/IFNγ. FP12 is known to affect TLR4 through interactions with MD-2 and cause internalization of CD14. In this regard, FP12 is thought to competitively exclude LPS from binding to MD-2 and cause internalization of CD14, further impairing the delivery mechanisms that aid delivery of LPS to the co-receptor. 15 The fact that FP12 inhibits LPS-induced cytokine induction even 30 min after LPS treatment of cells (Figure 4) suggests that FP12 can competitively inhibit ongoing signaling induced by LPS, presumably by displacing LPS from MD-2.
This suggested that FP12 can inhibit MyD88-Biased signaling after LPS has bound to TLR4/MD-2. FP12/LPS competition for MD-2 binding has been previously observed in binding experiments in vitro, 16 but the capacity of FP12 to interrupt TLR4 signaling after LPS treatment is a novel finding from this study. 16 Not only was FP12 an effective inhibitor of cytokine secretion in a human macrophage cell line, but we also confirmed our findings at the level of LPS-induced gene expression in primary murine macrophages (Figure 2).
Regarding toxicological studies similarly to FP7 MTT data confirmed that FP12 (0–10 μM) in the presence or absence of LPS did not affect THP-1 macrophage viability (Figure 1S). To extend these findings beyond the effects induced by LPS signaling, we also tested the efficacy of FP12 in a murine model of lethal influenza disease. We previously showed that therapeutic administration of Eritoran, FP7, and other TLR4 antagonists were protective in this model of acute lung injury.4,17 The data presented herein demonstrate that FP12 is at least as effective as FP7 in reducing lethality, lung histopathology, and cytokine gene expression. Eritoran cannot be used intraperitoneally because it interacts with high density lipoprotein in the peritoneal cavity (D.P. Rossignol, personal communication and data not shown) and for this reason has been administered intravenously. However, FP12 is effective whether administered i.v. (Figure 5(b)) or i.p. (Figure 6(a)), indicating another important benefit to this TLR4 antagonist.
The biological activity of compound FP12 and its mechanism of action is presented here. FP12 can efficiently block the TLR4/MyD88 axis even when administered after LPS thus reducing inflammatory cytokine production. Intraperitoneal administration of FP12 provides significant protection in mice infected with mouse-adapted influenza strain PR8 by suppressing influenza-induced ALI.
In our in vivo model of influenza-induced inflammatory disease, the cytokine storm causing animal lethality is largely mediated by HMGB1, a DAMP that has been implicated in the pathogenesis of multiple inflammatory diseases (e.g., sepsis, radiation injury, cardiovascular disease, arthritis, and others.30–32 Disulfide-linked HMGB1 is a TLR4 agonist that acts by binding to a site on MD-2 that is distinct from the LPS binding site. 9 P5779 is a small molecule inhibitor that binds to MD-2 and inhibits HMGB1-mediated binding and signaling, but not LPS-induced signaling. 9 Conversely, Eritoran inhibits both LPS- and HMGB1-mediated cytokine expression in macrophages. 2 Patel et al. showed that the levels of HMGB1 induced by infection of cotton rats with various human influenza isolates correlated well with the severity of disease; moreover, treatment of cotton rats with Eritoran blunted the levels of HMGB1. 12 A recent study showed that influenza infection results in significant lung tissue damage that can be ameliorated by treatment of mice with necroptosis inhibitors. 33 like Eritoran, FP12 reduced HMGB1-mediated cytokine gene expression in primary murine macrophages (Figure 3S) and significantly reduced the level of HMGB1 in lung homogenates of mice infected with PR8 (Figure 6(c)).
Overall, these results showed the potential of FP12 for further development in future preclinical and clinical studies for treatment of inflammatory-based diseases.
Supplemental Material
sj-docx-1-ini-10.1177_17534259241313201 - Supplemental material for Preclinical development of the TLR4 antagonist FP12 as a drug lead targeting the HMGB1/MD-2/TLR4 axis in lethal influenza infection
Supplemental material, sj-docx-1-ini-10.1177_17534259241313201 for Preclinical development of the TLR4 antagonist FP12 as a drug lead targeting the HMGB1/MD-2/TLR4 axis in lethal influenza infection by Kari Ann Shirey, Alessio Romerio, Mohammed Monsoor Shaik, David S Leake, Charys Palmer, Natalia Skupinska, Jules Paton, Grisha Pirianov, Jorge CG Blanco, Stefanie N Vogel and Francesco Peri in Innate Immunity
Footnotes
Abbreviations
Acknowledgments
The CINMPIS consortium in Advanced Organic Synthesis, Italy
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by The University of Maryland Baltimore, Institute for Clinical & Translational Research (ICTR) grant #1UL1TR003098 (SV, KAS, JP); British Heart Foundation (project grant PG/20/10072)(VA, DSL) (GP); PRIN 2022-TENET – Targeting bacterial cell ENvelope to reverse rEsisTance in emerging pathogens-Founded by European Community; EU- Next Generation EU, Missione 4 Componente 1 CUP H53D23004750006 (FP)- DD 104/2022; FAQC 2023– Grant from the University of Milano-Bicocca (FP); GP was supported by ARU PhD studentship.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.