
Abstract
Progressive HIV infection is characterized by profound enterocyte damage, microbial translocation and chronic immune activation. We aimed to test whether High Mobility Group Box protein 1(HMGB1), a marker of cell death, alone, or in combination with LPS, might contribute to HIV-associated immune activation and progression. Altogether, 29 untreated HIV-infected individuals, 25 inflammatory bowel disease (IBD) patients and 30 controls were included. HIV-infected patients had lower plasma LPS levels than IBD patients, but higher levels of soluble CD14 and Myeloid Differentiation (MD) 2, which interacts with TLR4 to initiate LPS-signalling. Furthermore, plasma levels of HMGB1 and MD2 were correlated directly within the HIV-infected cohort (r = 0.89, P < 0.001) and the IBD-cohort (r = 0.85, P < 0.001), implying HMGB1 signalling through the MD2/TLR4-pathway. HMGB1 and LPS, although not inter-correlated, were both moderately (r = 0.4) correlated with CD38 density on CD8+ T cells in HIV progressors. The highest levels of CD38 density and MD2 were found in progressors with plasma levels of both LPS and HMGB1 above the fiftieth percentile. Our results could imply that, in some patients, immune activation is triggered by microbial translocation, in some by cell death and in some by HMGB1 in complex with bacterial products through activation of the MD2/TLR4-pathway.
Keywords
Introduction
Chronic immune activation is a central feature of progressive HIV-1 infection and includes polyclonal B-cell activation, 1 increased T-cell activation,2,3 and increased levels of pro-inflammatory cytokines and chemokines. 4 Notably, the degree of immune activation measured by CD38+ CD8+ cells is a better predictor of disease progression and mortality than plasma viral load. 5
Beyond viral replication per se and opportunistic pathogens, HIV-induced T-cell depletion in the gastrointestinal (GI) tract and subsequent microbial translocation have been proposed to be the major drivers of HIV-1-associated immune activation. 6 A damaged GI tract leads to systemic translocation of luminal microbial products. These include pro-inflammatory trigger molecules, such as LPS, flagellin and bacterial CpG DNA—all which stimulate the innate immune system through TLR upon recognition of highly conserved pathogen-associated molecular patterns. 6
Several soluble proteins tightly regulate the signalling of LPS through the TLR4 pathway. First, LPS-binding protein extracts LPS from Gram-negative cell membranes or soluble translocated LPS, thus facilitating binding to the LPS cell surface receptor CD14. In order to activate TLR4, LPS requires binding by the Myeloid Differentiation 2 (MD2) complex, where CD14 plays a central role by transferring LPS to MD2.7,8 Upon LPS-binding, it is likely that MD2/TLR4 form heterodimers, which initiates the signalling cascade. 9 In subjects with progressive HIV-1 infection, increased LPS levels are associated with both elevation of activated CD38+ CD8+ T-cells and raised IFN-α levels. 10 Importantly, the classical LPS signalling TLR4 pathway cannot alone explain these findings, 10 suggesting the involvement of other factors, including translocation of other microbial trigger molecules.
High Mobility Group Box protein 1 (HMGB1) is a nuclear DNA binding protein with various properties, such as cell cycle regulation and gene expression through the condensing or unfolding of chromatin. 11 HMGB1 may be liberated from cells in two different ways: the protein is either secreted actively by intact cells upon pro-inflammatory stimuli or passively released from damaged cells into the extracellular milieu, in which it may act as an alarmin by signalling tissue damage and initiating reparative responses.12–15 Extracellular HMGB1 is also a potent pro-inflammatory protein, partly by forming complexes with various TLR ligands. 16 We have demonstrated recently that TLR ligands, together with HMGB1, synergistically induce replication of HIV in vitro. 17 Furthermore, we found that the combination of elevated LPS and HMGB1 in vivo was associated with high viral load, 18 indicating that each of these substances promote TLR-mediated immune activation in HIV-1 infection. 19 In fact, HMGB1 might even fuel the TLR4 pathway by binding specifically to LPS, facilitating disaggregation of LPS from bacterial membranes with subsequent binding to CD14. 20 However, HMGB1 might also interact with other TLR ligands, such as CpG-DNA and flagellin, and signal through other receptor systems, such as Receptor for Advanced Glycation End-products (RAGE).14,16
To date, few studies have explored the potential contribution of cell death to HIV-associated systemic immune activation. We hypothesized that HMGB1 alone, or in combination with LPS, might contribute to immune activation and disease progression in HIV-1-infected individuals not receiving antiretroviral therapy (ART).
Materials and methods
Study design and population
Baseline characteristics of the HIV-1-positive subjects (n = 28).

To contextualize our data, we included a group of healthy controls (n = 30) and a group of patients with inflammatory bowel disease (IBD; n = 25), in which microbial translocation and TLR dysfunction are thought to play pivotal roles in the pathogenesis. 22 The IBD patients (13 Crohn’s disease; 12 ulcerative colitis) were in remission and not HIV-infected: 19 of 25 received immune modulating treatment (of whom 17 received aminosalicylates, 10 received steroids, none received TNF inhibitors), 4 had been surgically treated and 4 received no treatment. Median age was 39 years [interquartile range (IQR) 29–45 years] and 15 of 25 were male. They were all Caucasian. It should be noted that the control group [median age 50 (43–55) years, 23 of 30 male, all Caucasian] and IBD group were not strictly matched with the HIV-1-infected cohort. Written informed consent was obtained from all subjects and the study was performed in accordance with the Declaration of Helsinki.
Material and analyses
Plasma samples were collected with standard venipuncture, separated and frozen within 30 min and stored at −80℃ until analysis. Pre-sampling fasting state was recommended, but not recorded. For HIV-infected individuals, freshly drawn EDTA venous blood was obtained and stained with combinations of fluorescence-labelled mAbs for determination of CD3, CD4, CD8 and CD38 (all Becton Dickinson, Oxford, UK). CD38 density was calculated per cell in the CD38+ subset by phycoerythrin-labelled Quantibrite anti-CD38 with a mAb:phycoerythrin labelling ratio at 1 : 1, using Quanibrite phycoerythrin-labeled beads (Becton Dickinson) to define a standard curve. Samples were analysed with flow cytometry (FACS Canto II with Diva software version 6.1; Becton Dickinson) as described previously. 23
HMGB1 (Shino-Test Corp., Oonodai, Kanawaga, Japan) and soluble CD14 (R&D, Minneapolis, MN, USA) were analysed by ELISA according to the manufacturer’s instructions. MD2 was analysed by in-house ELISA as described previously. 24
LPS was analysed by limulus amoebocyte lysate colormetric assay (Lonza, Walkersville, MD, USA) according to the manufacturer’s instructions, with the following modifications: samples were diluted 10-fold to avoid interference with background color and preheated to 68℃ for 12 min prior to analyses to dissolve immune complexes as described previously. 18
Intestinal fatty acid binding protein (I-FABP), a marker of enterocyte damage, was measured initially with a previously-described ELISA-kit (Hycult, Uden, the Netherlands). 25 However, unacceptably high background was obtained by us with the current batches (Trøseid et al., ICMI, Paris, 2011) and others (D. Douek, personal communication). Hence, we therefore re-analysed our samples with a recently developed ELISA for measurement of I-FABP. 26
To minimize the inter-assay variability, all of the soluble markers from progressors, non-progressors, IBD patients and controls were analysed in the same run.
Two subjects (one HIV-infected progressor and one IBD patient) were excluded from measurement of HMGB1 and LPS owing to visible haemolysis and background colour, which are known to interfere with the analyses.
Statistical analyses
All data are presented as median values (25–75 percentile range) unless stated otherwise. Non-parametric statistics were used throughout. Between-group differences were evaluated with Mann–Whitney U test. Correlation analyses were performed using the Spearman method. Trend analyses of continuous data were evaluated by Jonckheere–Terpstra test. Chi-square likelihood ratio was used for trend analyses of categorical data. A significance level of 0.05 was used. The statistical analyses were performed with SPSS software, version 19.0 (SPSS Inc., Chicago, IL, USA).
Results
Soluble biomarkers in HIV-1-infected individuals, IBD patients and controls
Plasma levels of LPS were higher in IBD patients compared with HIV-1-infected individuals (P = 0.010, Figure 1A), suggesting that microbial translocation is more pronounced in clinically stable IBD patients. Conversely, the HIV cohort had elevated levels of MD2 (P = 0.002, Figure 1B), I-FABP (P = 0.012, Figure 1C) and, possibly, sCD14 (P = 0.101, Figure 1D) compared with IBD patients, suggesting higher levels of ongoing enterocyte damage and TLR4 activation. HMGB1 levels were not significantly elevated in HIV-infected subjects (median 2.00, IQR 1.73–2.30 ng/ml) compared with IBD patients (median 1.75, IQR 1.20–2.68 ng/ml) (P = 0.639).
IBD patients have elevated LPS levels compared with HIV-infected patients, but lower levels of MD2, I-FABP and possibly sCD14. Plasma levels of (A) LPS, (B) MD2, (C) I-FABP and (D) sCD14 in HIV-infected individuals (n = 22) and IBD patients (n = 25). Boxplots show medians and interquartile range with tenth and ninetieth percentiles; white circles are outliers. I-FABP, intestinal fatty acid binding protein; sCD14, soluble CD14.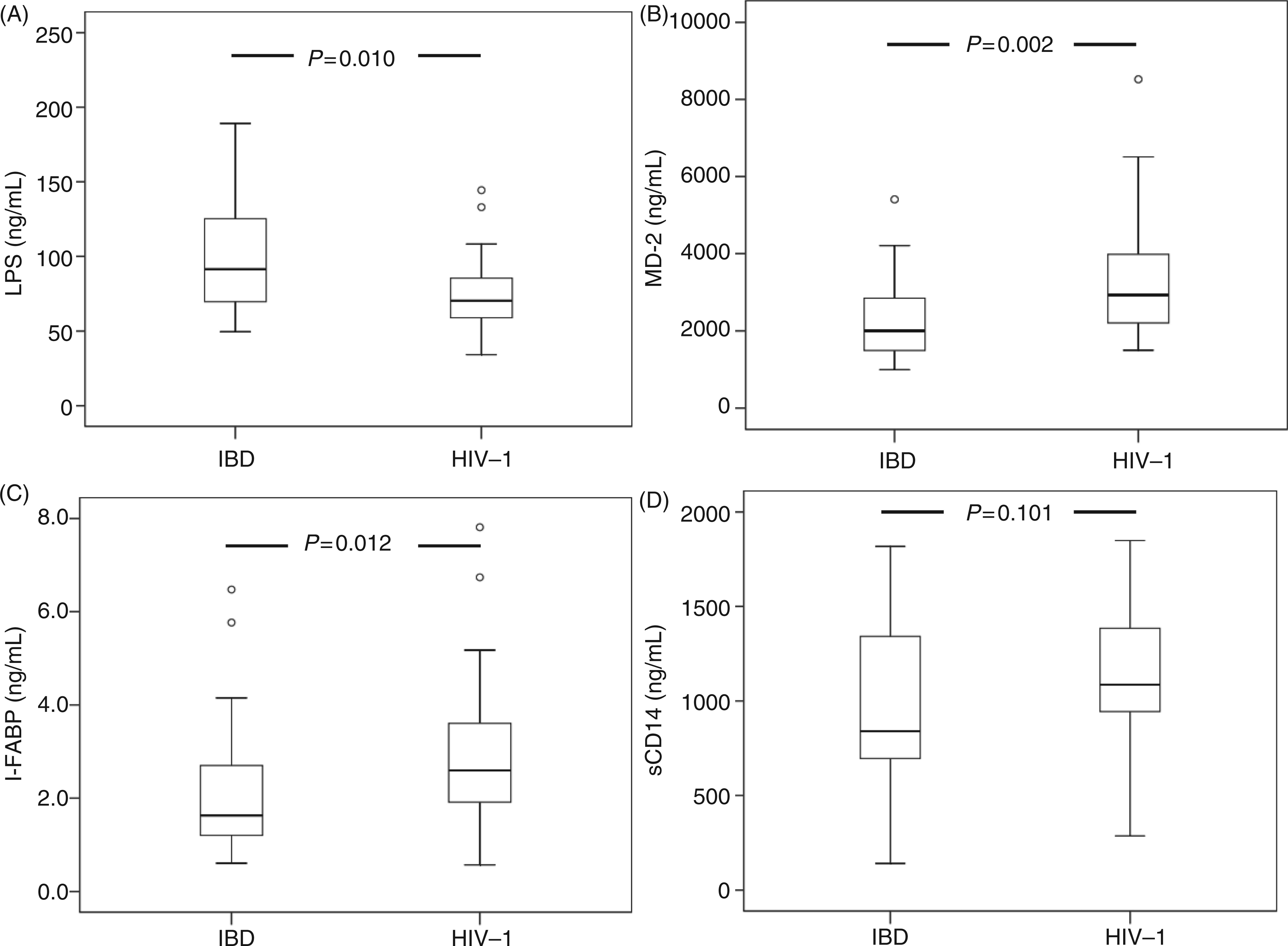
Compared with healthy controls, IBD patients and HIV-infected individuals had higher plasma levels of HMGB1 (P < 0.001 for both), LPS (P < 0.001 for both) and sCD14 (P = 0.006 and P < 0.001, respectively), whereas MD2 was elevated significantly only in HIV-infected individuals (P < 0.001). In healthy controls, median HMGB1 levels were 0.81 (IQR 0.49–1.38 ng/ml) and MD2 1860 (1104–2106 ng/ml), whereas LPS and sCD14 levels were as given in Figure 2. I-FABP was not measured in controls owing to lack of available ELISA kits.
Ulcerative colitis, Crohn’s disease and HIV-infection have different patterns of microbial translocation. Plasma levels of (A) LPS and (B) sCD14 in healthy controls (control; n = 30), ulcerative colitis (UC; n = 12), Crohn’s disease (Crohn; n = 13), HIV-infected progressors (Prog; n = 22) and long-term non-progressors (LTNP; n = 7).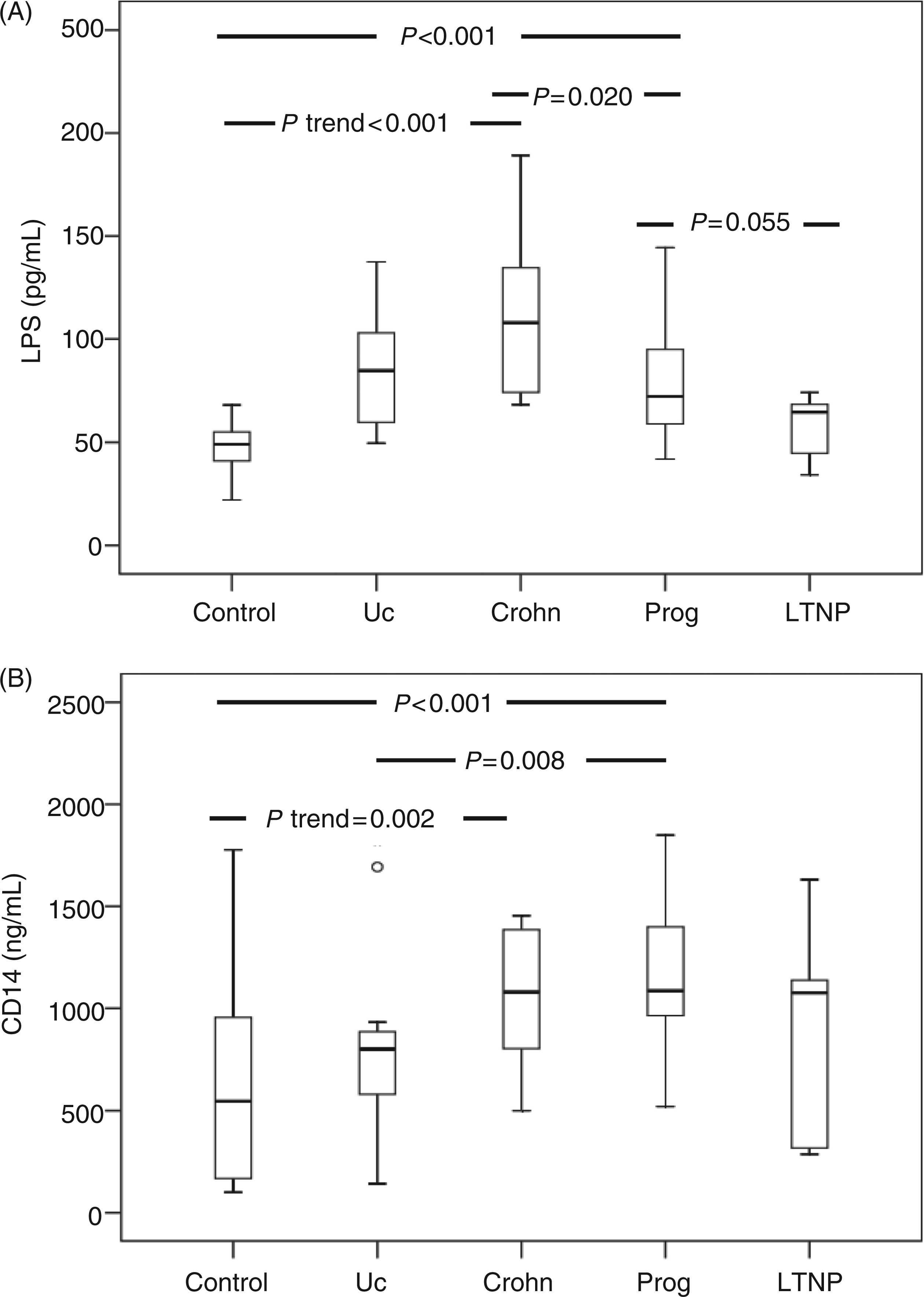
No significant differences were found in age and gender distribution between the IBD and HIV cohort, whereas the control group was somewhat older than the IBD and HIV cohorts (P ≤ 0.001). However, age, gender or ethnicity were not associated with any of the soluble biomarkers in any of the disease categories (data not shown).
Within the IBD cohort there were no significant differences in any of the soluble biomarkers between medically or surgically treated and untreated patients (data not shown), in line with a report showing no difference in systemic bacterial DNA levels between medically treated and untreated IBD patients. 27 However, when comparing healthy controls, ulcerative colitis and Crohn’s disease separately, there was a trend of increasing levels of LPS (P < 0.001, Figure 2A) and sCD14 (P = 0.002, Figure 2B), whereas no differences where seen for MD2, I-FABP or HMGB1 between Crohn’s disease and ulcerative colitis (data not shown).
Within the HIV-infected cohort, progressors had higher LPS levels compared with non-progressors (P = 0.055, Figure 2A). In contrast, there were no differences in sCD14, MD2, I-FABP or HMGB1 levels between progressors and non-progressors (data not shown). Plasma levels of MD2 and HMGB1 were strongly correlated within both the HIV-infected cohort (r = 0.89, P < 0.001) and the IBD cohort (r = 0.85, P < 0.001) (Figure 3), in contrast to LPS, sCD14 and I-FABP, which were neither inter-correlated nor correlated with MD2 and HMGB1 (data not shown).
MD2 is closely correlated with HMGB-1 in HIV-infected and IBD patients. Plasma levels of MD2 and HMGB-1 in HIV-infected (black circles) and IBD patients (white circles).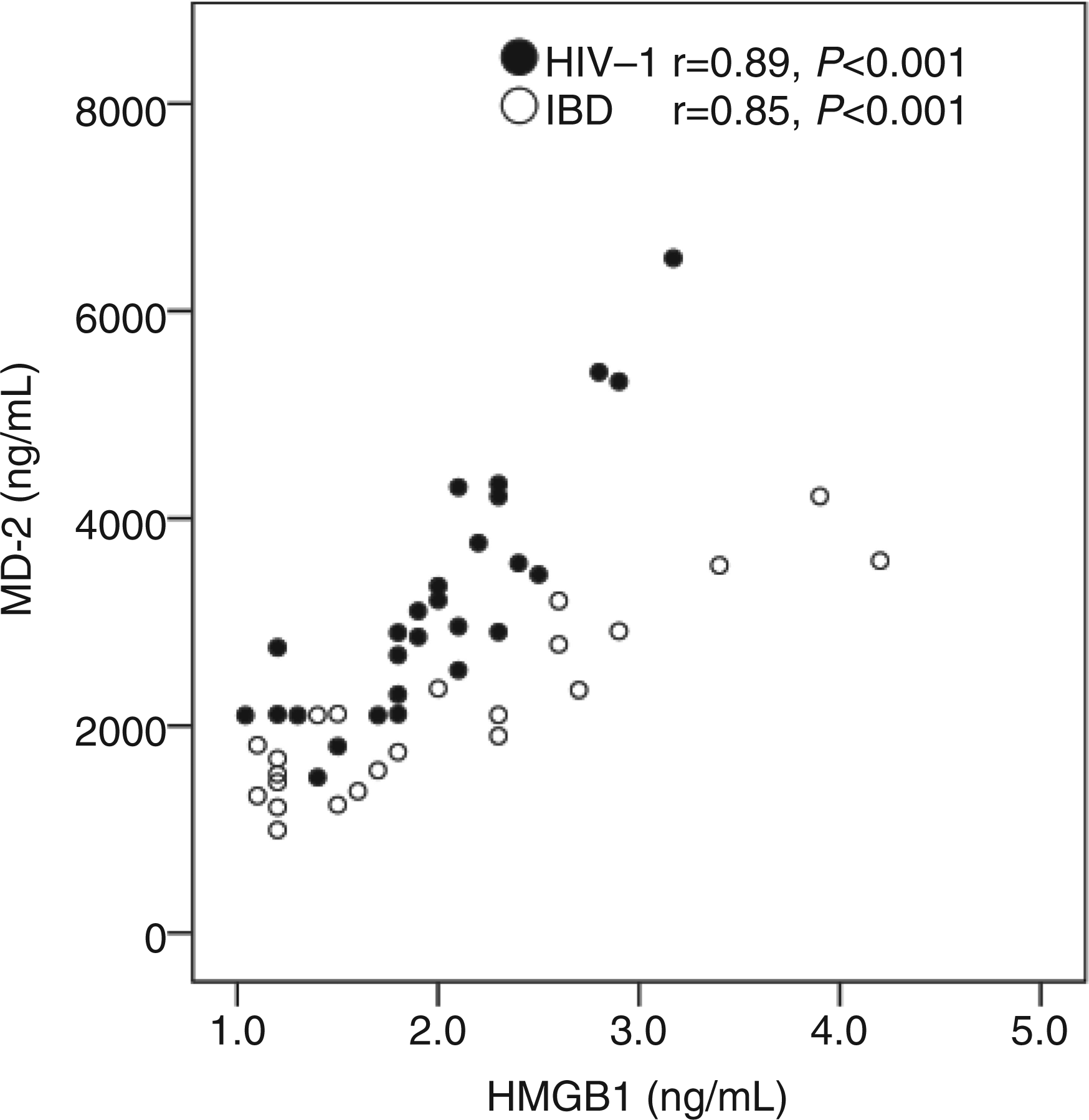
Plasma levels of LPS and HMGB1 correlate separately with CD38 on T cells in HIV progressors
Correlation analyses regarding immune activation and disease progression were performed in HIV-1 infected progressors (n = 22), the study group of main interest. In the progressors, plasma levels of LPS and HMGB1 were both correlated positively with the CD38 density on CD8+ T cells (r = 0.43, P = 0.047; r = 0.44, P = 0.044 respectively), but they were not inter-correlated, in line with our previous observation. 18 However, plasma MD2 did not correlate significantly with the chronic activation marker CD38 (r = 0.33, P = 0.147), despite its strong correlation with HMGB1. Moreover, CD38 did not correlate with either sCD14 (r = 0.24, P = 0.310) or I-FABP (r = 0.10, P = 0.678).
Interacting effect of LPS and HMGB1 on immune activation in HIV progressors
We have shown previously that circulating levels of LPS and HMGB1 have interacting effects on viral replication in vitro
17
and in vivo.
18
To further explore the potentially interacting effects of these two pro-inflammatory mediators on chronic immune activation, HIV-infected progressors were divided according to the fiftieth percentile of HMGB1 and LPS respectively. Three distinct groups were defined by having both markers below the fiftieth percentile (LoLo, n = 5), one marker elevated (HiLo, n = 11) or both markers elevated (HiHi, n = 5). Trend analyses throughout these strata showed increasing density of CD38 on CD38+ CD8+ T cells (P = 0.006, Figure 4A), increasing MD2 levels (P = 0.002, Figure 4B), lower CD4+ counts (P = 0.040), possibly higher viral loads (P = 0.095) and an increased tendency to ART initiation one year post-study (P = 0.082).
The impact of HMGB1 and LPS on immune activation. Potentially interacting effect of HMGB1 and LPS on (A) activated T cells (CD38 density) and (B) MD2. Trend analysis refers to comparison of both LPS and HMGB1 below the fiftieth percentile (LoLo, n = 5), one marker elevated (HiLo, n = 11) or both markers elevated (HiHi, n = 5). Boxplots show medians and interquartile range with tenth and ninetieth percentiles.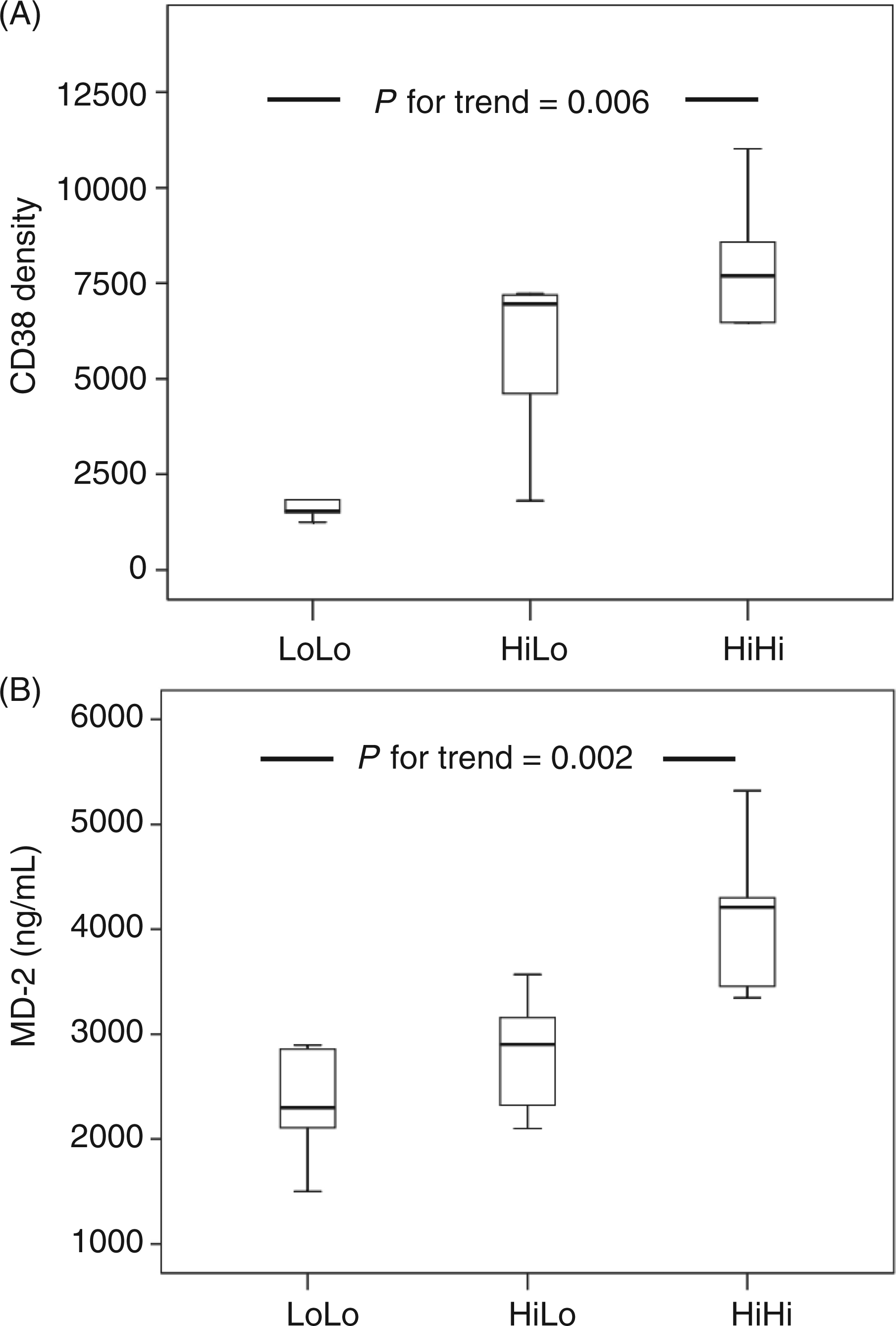
Notably, neither LPS, HMGB1, MD2 nor any of the soluble markers alone were associated significantly with current CD4 count, viral load or ART initiation among the HIV-infected progressors.
Discussion
In the present study, the close correlation between HMGB1 and MD2 both in HIV-1 and IBD is a novel finding, which supports the possibility that HMGB1 is a key trigger of the MD2/TLR4 pathway. Our results are well in line with previous reports, which show that HMGB1 might signal through,28,29 and even bind specifically, to TLR4. 30 Furthermore, HMGB1 might enhance cytokine production by forming complexes with LPS and thereby increase TLR4 activation. 31 Notably, we found that MD2 levels were highest in the patients who had elevated levels of both HMGB1 and LPS (HiHi), suggesting interacting, and possibly additive, effects of the two markers on the MD2/TLR4 pathway.
MD2 is either secreted directly as a soluble, active protein or may bind directly to TLR4 on the cell membrane. On the cell membrane, MD2 plays a key role by recognizing LPS and forming a heterodimer with TLR4, which is probably necessary to initiate the signalling cascade.24,32 However, limited amount of data are available on the cellular sources and function of soluble MD2. Nevertheless, elevated soluble MD2 levels have been demonstrated previously in HIV-infection using the same in-house ELISA kit as in the present study. 24 Furthermore, MD2 expression on monocytes was enhanced by IL-10, but not by IL-6 or TNF-α. 24 Although soluble MD2 is elevated in HIV-infection, MD2 per se is not necessarily harmful. In fact, excess MD2 might block LPS-responses in vitro 33 and it has been proposed that MD2 might have a dual role by participating in both the initiation and regulation of LPS-signalling through TLR4. 32
Another novel finding is that plasma levels of LPS and HMGB1 were both correlated positively with HIV-associated T-cell activation in terms of CD38 density on the CD8+ T cells without being inter-correlated. This is in accordance with our previous report that the combination of elevated LPS and HMGB1, although not inter-correlated, is associated with high viral load in HIV-infected patients. 18 As discussed previously, the association between LPS and immune activation cannot entirely rely on the TLR4 pathway. 10 Our findings suggest that cell death and the alarmin HMGB1 could be additional contributors to HIV-associated immune activation in terms of CD38, although the correlations were moderate.
In addition to trigging the TLR4 pathway, HMGB1 can potentially contribute to immune activation in HIV-infected patients by interacting with various TLR ligands, 16 as well as other receptor systems. For the RAGE pathway, a similar synergistic effect between HMGB1 and microbial products has been proposed. 34 Very recently, it was even shown that HMGB1 may promote recruitment of inflammatory cells by forming complexes with chemokines and signalling through the CXCR4 receptor, 35 which also functions as a co-receptor for HIV-1 on CD4+ T cells. 36 Furthermore, HMGB1 has been reported to be expressed at the synapsis between NK cells and dendritic cells, hence promoting early and widespread viral dissemination and latency by dendritic cells.37,38
The increased circulating HMGB1 levels found in the present study of untreated HIV-infected individuals could be accounted for by several cell types, as HMGB1 is present in all nucleated cells. 16 For example, new CD4+ T cells are infected constantly and die from infection or HIV-specific cytotoxic CD8+ T cells. This process, along with a generalized hyperactivated state, lead to premature death of T cells, which is compensated by a substantial enhanced recruitment and turnover of both the CD4+ and CD8+ subsets. 39 Proposed enhanced cell death and turnover of enterocytes 6 could also contribute to circulating HMGB1 in these patients. Although necrotic cells have been proposed to be the main source of systemic HMGB1, we found recently that both necrosis and apoptosis contribute to HMGB1 liberation during HIV-1-induced cell death, and that this alarmin could induce TNF-α release from peripheral blood mononuclear cells. 40 In addition to passive release from dying cells, HMGB1 is secreted actively from intact cells of the innate immune system upon various pro-inflammatory stimuli, including LPS. 41,42
It has been proposed that the intestinal barrier defect in HIV infection may resemble that of IBD. 43 However, our data suggest that although LPS levels were higher in IBD patients than in HIV-1-infected subjects, the consequences are different. IBD patients had lower levels of MD2, sCD14 and I-FABP, suggesting less immune activation and ongoing gut damage. This is in line with a study comparing patients with active IBD and HIV-1 reporting elevated LPS levels in both conditions, whereas only HIV-infected patients had significant elevation in activated CD8+ CD38+ T-cells and NK cells. 44 Interestingly, patients with Crohn’s disease had substantially higher LPS, but similar sCD14, levels compared with HIV-infected patients. Of note, sera from patients with active IBD have been proposed to facilitate LPS-signalling in intestinal epithelial cells via MD2 activity, in particular in Crohn’s disease. 45
Our data and the literature discussed earlier suggest that although microbial translocation and TLR activation are present in IBD 22 and in HIV infection, 6 different pathogenic mechanisms are probably at play. The early massive loss of sub-activated CCR5+ CD4+ Th17 cells in HIV-infected individuals 46 probably contributes to local and systemic immune activation, as well as HIV-specific loss of mucosal integrity. 6 Furthermore, proposed virotoxic effects of HIV on enterocytes 6 could account for the increased I-FABP levels observed in the HIV-infected progressors compared with the IBD cohort.
Our study has some limitations. Owing to lack of matching between the HIV-infected cohort, IBD patients and controls, the results should be interpreted with caution. Furthermore, although a fasting state was advised, but not formally recorded, post-prandial hypertriglyceridemia could have influenced the LPS-levels. 47 Moreover, the relatively small sample size increases the vulnerability for type II statistical errors. However, type I errors are less probable and the strong, significant correlation between HMGB1 and MD2 is thus likely to be reliable. Our study also has several strengths, including the prospective design and use of novel circulating markers allowing for in depth exploration of the TLR4 pathway in vivo.
In conclusion, plasma levels of HMGB1 and MD2 were strongly inter-correlated, with the highest levels of MD2 found in HIV progressors with elevated levels of both LPS and HMGB1. We propose that HIV-associated immune activation in some patients is triggered by microbial translocation, in some by cell death and, possibly, in some by the alarmin HMGB1 in complex with bacterial products through activation of the MD2/TLR4-pathway.
Footnotes
Acknowledgements
We thank the patients for their participation. We acknowledge the skilled assistance of the staff at the Outpatient Clinic of Infectious Diseases at Oslo University Hospital. All the authors contributed in planning the study and in critically reviewing and approving the final version of the manuscript. This work was supported by Oslo University Hospital and University of Oslo.
Conflict of interest
The authors declare that they have no conflict of interest.
Funding
This work was supported by Research council of Norway (grant no. 191514/S50 GLOBVAC) and Swedish Society for Medical Research.