
Abstract
Neutrophils constitute the first line of defense in human immunity and can be attracted to inflamed and infected sites by various chemokines. As essential players in immune processes, neutrophils theoretically play integral roles in the course of chronic inflammation-induced atherosclerosis. However, because neutrophils are rarely found in atherosclerotic lesions, their involvement in the pathophysiological progression of atherosclerosis has been largely underestimated or ignored. Recent research has revealed convincing evidence showing the presence of neutrophils in atherosclerotic lesions and has revealed neutrophil contributions to different atherosclerosis stages in mice and humans. This review describes the underlying mechanisms of neutrophils in different stages of atherosclerosis and highlights potential neutrophil-targeted therapeutic strategies relevant to atherosclerosis. An in-depth understanding of neutrophils’ roles in atherosclerosis pathology will promote exploration of new methods for the prevention and treatment of atherogenesis and atherothrombosis.
Introduction
Atherosclerosis has existed for at least 4000 years. 1 With increased human life expectancy, unhealthy diet habits, and lack of exercise and sleep, atherosclerosis ranks among the most common causes of morbidity and mortality among various lethal diseases. The essential characteristics of the disease were identified at the pathological level one hundred years ago; for example, the invasion of white blood cells, the formation of foam cells, and the accumulation of fat in blood vessels were established. 2 However, the mechanism of atherosclerosis pathogenesis is still unclear.
Several kinds of cells, which have been clearly identified over the years, are involved in atherosclerosis; these cells include vascular endothelial cells (ECs), vascular smooth muscle cells (SMCs), monocytes, macrophages and T cells.3,4 However, neutrophils, the most abundant of the white blood cells in the circulatory system, have received little attention in studies on atherosclerosis pathophysiology. Although a series of reports have indicated that neutrophils engage in crosstalk with monocytes, regulate the proinflammatory response of macrophages and modulate thrombogenesis, the role of neutrophils in the atherosclerotic process has not received meaningful attention.4–6 This lack of interest is largely due to the lack of specific detection methods, the relatively short lifespan of neutrophils, and, ultimately, the ability of neutrophil to undergo phenotypic changes and thus display markers that make them appear to be dendritic cell-like or macrophage-like cells.7–9 Nevertheless, recent reports have revealed different roles for neutrophils in atherogenesis.10–12 Thus, in this review, we summarize the latest findings on the role played by neutrophils in atherosclerosis. Furthermore, we pay special attention to neutrophil mechanisms of action and their importance in different atherosclerosis stages.
Mechanisms of atherogenesis
Currently, the commonly accepted theory on the pathogenesis of atherosclerosis is as follows (Figure 1): The initial events in the progression of atherosclerosis are endothelial injury and dysfunction, which cause the retention of apoB-containing lipoproteins (low-density lipoprotein cholesterol:LDL-C; very low-density lipoprotein cholesterol: VLDL-C; and apoE remnants) in the subendothelial space. Then, these lipoproteins are modified (oxidation, glycation, enzymatic) in pathologic states. oxidized LDL (Ox-LDL) increases the abundance of cell adhesion molecules, including vascular cell adhesion molecule-1 (VCAM-1), intercellular cell adhesion molecule-1 (ICAM-1), and P- and E-selectins, on ECs, leading to leukocyte (mainly T lymphocyte and monocyte) recruitment into the subendothelial space. These inflammatory cells migrate to the intima via the interaction of chemotactic proteins such as eotaxin, monocyte chemoattractant protein-1, and interferon-γ. Then, monocytes differentiate into macrophages and express scavenger receptors) such as CD 36 that take up Ox-LDL. These lipid-loaded macrophages are also called foam cells, which are hallmarks of early atherosclerotic lesions. Mast cells and T lymphocytes also migrate into the intima and along with foam cells release a variety of proinflammatory cytokines, reactive oxygen species (ROS) and growth factors to stimulate collagen deposition and SMC migration. SMCs in the tunica migrate into the intima in response to mediators released by accumulating leukocytes. During the development of atherosclerotic plaques, stimulated SMCs produce extracellular matrix molecules (such as elastin, glycosaminoglycans and proteoglycans) that contribute to the thickening of the intimal layer. Advanced atherosclerotic plaques are filled with cell debris, extracellular lipids, and foam cell, whereas stable plaques are protected from the circulating blood by fibrous caps. Plaque destabilization can be caused by proteolysis of fibrillar collagens. Foam cell–EC interaction increases the production of matrix metalloproteinases (MMPs), which degrade the extracellular matrix in the fibrous cap of each atherosclerotic plaque and at the basement membranes of ECs, resulting in physical plaque disruption and thrombus formation.13,14
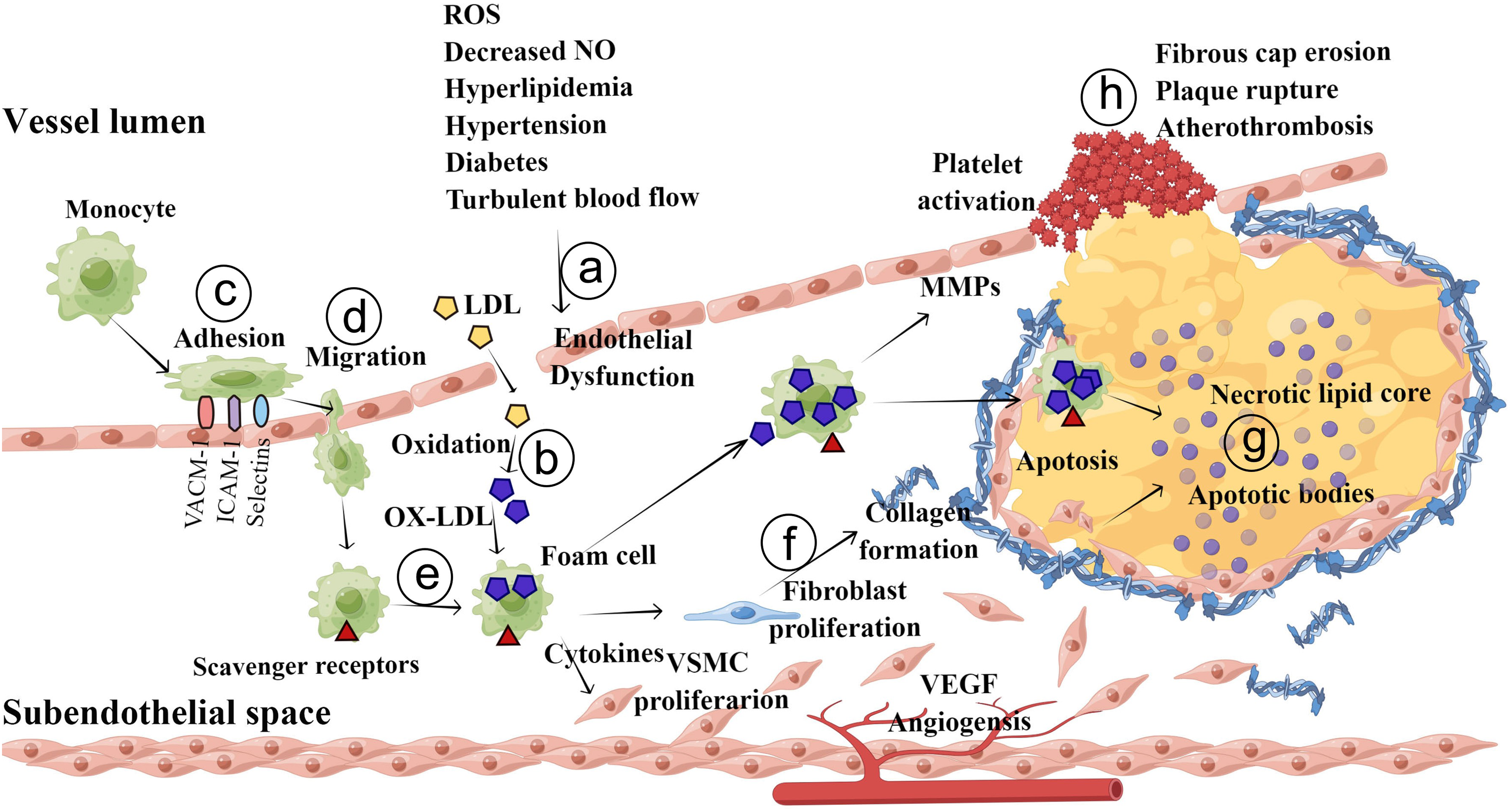
Pathogenesis of atherosclerosis. a, The fatty streak phase of atherosclerosis begins with dysfunctional ECs and the retention of lipoproteins (LDL, VLDL, etc.) in the subendothelial space. b, Retained lipoproteins are modified (via oxidation, glycation, and enzymatic modification). c, The modified lipoproteins (mainly Ox-LDL), along with other atherogenic factors, promote activation of ECs and increase the expression of selectins, VCAM-1 and ICAM-1, leading to attachment of monocytes and other immune cells. d, These adherent monocytes and other immune cells migrate into the intimal space. e, Monocytes in the intimal space take up Ox-LDL and differentiate into foam cells. f and g, Foam cells and immune cells release a variety of cytokines, growth factors and ROS to stimulate SMCs migration and collagen deposition, leading to the development of an atheromatous plaque. h, In the advanced stage of atherosclerosis, upregulated MMPs destroy the fibrous caps of atherosclerotic plaques and the basement membranes of ECs, resulting in physical plaque disruption and thrombus formation.
Neutrophil profiling and identification
Neutrophils are formed from hematopoietic stem cells in the bone marrow. Approximately109 to 1010 mature neutrophils are released each day from marrow in response to granulocyte colony-stimulating factor and other stimuli, and as the most abundant type of granulocyte, makes up 40% to 70% of all white blood cells. The average lifespan of inactivated human neutrophils in the circulatory system has been estimated via different approaches to be between 5 and 135 h.15,16 Neutrophils can be activated and migrate to tissues, where they survive for 1–2 days. Neutrophils are also released into the blood from a splenic reserve following myocardial infarction. 17 Neutrophils undergo chemotaxis via amoeboid movement, which allows them to migrate toward sites of infection or inflammation. Cell surface receptors enable neutrophils to detect chemical gradients of molecules such as IFN-γ, interleukin-8 (IL-8) and leukotriene B4, which neutrophils use to forge a migration path. Neutrophils play a crucial role in the front-line defense against invading pathogens via three processes: phagocytosis (ingestion), degranulation, and generation of neutrophil extracellular traps (NETs). 18 Brinkmann et al. first described NETs release as a particular combination of apoptosis and necrosis that releases condensed chromatin and antimicrobial proteins. 19 However, NETs formation differs from typical apoptosis and necrosis. 20 Neutrophils undergo histone citrullination via an enzyme called protein arginine deiminase 4 (PAD4), which converts arginyl residues in histones (particularly H3 and H4) to citrulline, breaking ionic bonds that usually prevent DNA from leaving nucleosomes. 21 This release of ionic bonds leads to the dissociation of linking histones and heterochromatin from the nucleosome structure, and eventually, the nuclear chromatin network is released into the extracellular space and the surrounding tissue.
Neutrophils are double-edged swords. On the one hand, they cause tissue damage due to their infiltrative behavior and anti-inflammatory byproducts. In particular, tissue damage and the promotion of atherosclerosis and thrombosis via NETs have received increasing attention.12,22 On the other hand, they facilitate wound repair via a series of mechanisms. 23 Neutrophils can regulate macrophage behavior, display innate immune memory, and play an essential role in the course of atherosclerosis.24,25 Nevertheless, due to their short lifespan and the rapid removal of senescent neutrophils by macrophages, failure to identify neutrophils accurately and precisely limited the understanding neutrophil roles in atherosclerosis for a long time.
The emergence of neutrophil-specific antibodies enabled accurate and effective identification of neutrophils, even in small amounts, in tissues. Marcella et al. first described the distribution of neutrophils in atherosclerotic plaques in mice by using the neutrophil-specific mouse monoclonal antibody Ly-6G in 2008. 26 In early murine lesions, Ly-6G-labeled neutrophils were identified in subendothelial and intimal locations. In more advanced plaques, neutrophils were found in the plaque shoulder and the adventitia. 27 Two years later, in humans, CD-66b-labeled neutrophils were found in different regions of plaque, with high numbers in the fibrous cap, the shoulder, the plaque–medium interface, and in areas with intraplaque bleeding. Lower numbers of neutrophils have been found underneath the luminal endothelium or in the vicinity of microvessels in a plaque. 28 In addition, single-cell RNA sequencing has contributed to characterization of neutrophils in human and murine atherosclerotic lesions.29,30
Neutrophil transendothelial migration and endothelial dysfunction
The endothelium is a semipermeable monolayer of ECs connected by tight junctions that separates all tissues from circulating blood. The vascular endothelium is a highly active organ involved in the regulation of cell coagulation, adhesion and migration, vascular tone, vessel wall permeability, and various inflammatory processes. 31 Endothelial dysfunction is considered the earliest detectable and reversible stage of atherosclerosis. Increasing evidence indicates that neutrophil adhesion across ECs is closely related to EC dysfunction.
Neutrophil transendothelial migration is a tightly regulated process in which cells roll along a vessel wall, subsequently adhering to the EC surface and migrating across the endothelium. This process is mediated by adhesion molecules with expression that is rapidly upregulated by inflammatory cytokines, including tumor necrosis factor (TNF-α) and Interleukin 1β (IL-1β). 32 In particular, the attachment of neutrophils to P-selectin and E-selectin expressed by ECs slows neutrophils and enables them to roll under relatively high shear stress.33,34 Subsequently, firm adhesion is ensured by the binding of neutrophil CD11/CD18 integrins to endothelial adhesion molecules. Migration proceeds through either the paracellular route via EC‒cell junctions 35 or through the transcellular pathway via the EC body.36,37 Neutrophil transendothelial migration causes direct physical damage to the integrity of ECs and endothelial dysfunction due to the production of ROS, secretion of inflammatory mediators and neutrophil degranulation. 38 ROS can induce vascular hyperpermeability, due to junction disruption and EC contraction, via endothelial NLRP3 inflammasome 39 and myosin light chain kinase activation. 40 Moreover, neutrophils can release TNF-α, IL-1β, and the chemokines CXCL1, 2, 3, and 8 to increase the permeability of ECs.41,42 In addition, neutrophil-derived Leukotriene A4 induces the synthesis of biologically active Leukotriene B4. Leukotriene B4 then activates neutrophils to release heparin-binding protein (a granule component), causing EC contraction. 43 Through degranulation, neutrophils release elastase, metalloproteases and cathepsin G (CTSG), which can breakdown glycocalyx constituents, junctional complexes, and focal adhesion components.44,45 NETs are mainly composed of DNA, histones, and the contents of certain granule components in neutrophils. Previous studies have suggested that NETs are very effective means by which neutrophils kill invading pathogens before neutrophil death. 46 An increasing number of studies have confirmed that NETs exhibit a variety of other biological functions. 47 Adhesion of NETs to the EC surface may contribute to EC dysfunction. Citrullinated histone H3, a major protein component of NETs, causes microvascular leakage and barrier dysfunction by disrupting adherens junctions and rearranging the contractile cytoskeleton in ECs. 48 Jason et al. found that preincubation of mouse aortic rings from 8-week-old NZM mice with NETs significantly impaired vasorelaxation in response to acetylcholine. 49 In contrast, Cl-amidine, which blocks NETs release from neutrophils, was used to treat aortic rings, leading to the formation of fewer NETs and causing less endothelial damage. 49 Carmelo et al. confirmed that MMP9 in NETs activated MMP2 on ECs, resulting in EC dysfunction. Moreover, inhibition of MMP2 activation decreased NETs-induced vascular cytotoxicity and restored endothelium-dependent function. 5
Neutrophils and LDL
The uptake of LDL by ECs and subsequent oxidative modification is characteristic of early atherosclerosis. Human α-defensins, also called human neutrophil peptides, play essential roles in the development of early atherosclerosis. 50 α-Defensins are antimicrobial proteins that constitute 5% of the total protein in neutrophils and are released from a subset of neutrophils that are activated by various cytokines. 51 α-Defensins are found in human carotid arteries and atherosclerotic coronary arteries.52,53 A significant correlation has been identified between the deposition of α-defensins in skin tissue and the severity of coronary artery disease. 54 Defensin and LDL in solution and on endothelial surfaces formed stable complexes. Interestingly, defensin promoted the enhancement of LDL binding to heparan sulfate-containing proteoglycans but not to the LDL receptor.55,56 Higazi et al. studied the effect of α-defensins on LDL trafficking, metabolism and atherogenesis using human α-defensin-expressing neutrophils in transgenic mice. They found that α-defensin-expressing mice when fed a regular diet and with normal plasma levels of LDL formed α-defensin/LDL complexes, accelerated the clearance of LDL from the circulatory system, and enhanced vascular deposition and retention of LDL via a posttranslational modification of LDL (Figure 2). Additionally, α-defensins induced endothelial cathepsin expression, increased endothelial permeability to LDL, and accelerated the development of lipid streaks in the aortic roots in the same mice treated as described above. Transplantation of bone marrow from α-defensin-expressing mice to wild-type (WT) mice increased LDL clearance, increased vascular permeability, and expanded vascular deposition of LDL. However, transplantation of WT bone marrow into α-defensin-expressing mice prevented these outcomes, and the same results were obtained when α-defensin-expressing mice were treated with colchicine to inhibit the release of α-defensins. 50

Neutrophils promote LDL vascular deposition and oxidation during the early stage of atherosclerosis. a, Defensin and LDL form stable complexes that bind to the endothelial surface. b, α-Defensins promote endothelial cathepsin expression and increase endothelial permeability to LDL. c, Activated neutrophils oxidize LDL to form Ox-LDL, releasing ROS, which induce endothelial damage. d, Ox-LDL induces NETs formation via TLR-PKC-IRAK-MAPK and NADPH oxidase activation. e, NETs release more ROS that in turn oxidize LDL to Ox-LDL.
Neutrophils are closely related to the oxidative modification of LDL. Ox-LDL is a crucial initiator described in Steinberg's“oxidative theory of atherosclerosis”. 57 Ox-LDL activates the endothelium by stimulating the expression of adhesion molecules, the recruitment of mononuclear cells to the intima, the transformation of monocyte-differentiated macrophages into foam cells, and the accumulation of foam cells to form fatty streaks. Neutrophils produce ROS when rolling or adhering to ECs. 58 The oxidative modification of LDL by ROS has been extensively investigated.59,60 However, Ox-LDL induces neutrophil NETs formation via the TLR-PKC-IRAK-MAPK pathway and NADPH-oxidase activation, which means neutrophils undergo apoptosis and release more ROS. 61
Neutrophil and monocyte recruitment
Monocytes and monocyte-derived macrophages are abundant in atherosclerotic plaques, and their importance in atherosclerosis is widely acknowledged. 30 However, a study on enzymatically digested aortas from neutropenic mice and WT mice fed a high-fat diet revealed reduced numbers of classical monocytes and macrophages in the aortas of the neutropenic mice compared to the WT mice, suggesting that neutrophils contribute to the accumulation of monocytes and monocyte-derived macrophages in lesions. 62 Notably, the loss of macrophages also reduced the uptake of neutrophil apoptotic bodies. Inefficient removal of apoptotic cells by macrophages may lead to the formation of necrotic cores by inducing postapoptotic necrosis and accumulation of necrotic debris. 63 An increasing number of studies have indicated that neutrophils contribute to monocyte recruitment and adhesion to ECs. Schumski et al. reported that inflammation triggered the deposition of NETs on activated ECs along the arterial lumen and that the NET-associated histone H2a mediates charge-dependent monocyte adhesion to ECs and accelerates atherosclerosis. 64 In addition, monocytes and neutrophils can release CCL2, which then accumulates in ECs and mediates the adhesion of monocytes and neutrophils to ECs. 65 Of note, activated ECs can stimulate adherent neutrophils to produce more NETs, which allows more monocytes to adhere to ECs. 66
CTSG is stored in neutrophil azurophil granules and secreted upon neutrophil activation. Soehnlein et al. found that monocytes suspended in CTSG-free conditions showed hampered adhesion capacity when incubated with monocytes from WT mice with plasma derived from either Apoe–/– or Apoe–/–Ctsg–/– mice fed a high-fat diet for four weeks, and adhesion to TNF-activated ECs was evaluated. 67 Interestingly, CTSG controlled the recruitment of monocytes only in arteries, not venula, because the recruitment of CTSG to monocytes proceeds under artery-like shear stress between 10 and 40 dyn/cm2, and the shear stress of postcapillary venules is lower. This regulated process must involve platelets. Moreover, therapeutic neutralization of CTSG specifically abrogated arterial monocyte adhesion without affecting monocyte adhesion in the microcirculation, and intervention with a CTSG neutralizing antibody inhibited atherogenesis in mice. 67 Cathelicidin, another neutrophil-secreted protein, induced firm adhesion of classical CD14++CD16− monocytes to ECs. In cathelicidin-deficient (Cramp−/−) mice, the recruitment of classical monocytes by ECs after TNFA stimulation was significantly reduced. Cathelicidin induced the recruitment of classical monocytes by activating formyl-peptide receptor 2 on the surface of monocytes in the basal-apical direction across the endothelium. Formyl-peptide receptor 2 activation mediated firm adhesion through inside-out integrin signaling and subsequent changes in integrin conformation toward an extended, high-affinity state. 6 Additionally, azurocidin, deposited on the endothelium by activated neutrophils, has been shown to induce monocyte extravasation via FPRs, thereby stimulating monocyte adhesion. 68 Azurocidin and related granule proteins further favor the extravasation of classical monocytes by stimulating chemokine release from monocytes and macrophages. 69 In contrast to these mechanisms, neutrophil-derived serine proteases activate and enhance the chemotactic activity of chemokines on monocytes by cleaving chemokines at serine residues. 70 Many cytokines, chemokines, and their respective receptors contain cleavage sites for neutrophil serine proteases. For example, N-terminal cleavage of IL-8 by proteinase-3 and neutrophil-activating protein-78 (CXCL5) via CTSG releases truncated forms of these chemokines that show higher chemotactic activity than that of the full-length molecules.71,72 Similarly, N-terminal modification of CCL15 increases its monocyte chemotactic activity many-fold via CTSG.73,74
Neutrophils modulate macrophage phenotype and foam cell formation
Driven by macrophage colony-stimulating factors and probably other differentiation factors, most monocytes in early atherosclerotic lesions become cells with macrophage-like features. 75 Macrophages are essential players throughout all stages of atherosclerosis. 75 The interest in macrophage heterogeneity in atherosclerotic lesions has been profound, particularly with respect to macrophages involved in proinflammatory processes (M1 macrophages) versus those involved in resolution and repair (M2 macrophages). Nevertheless, a clear picture has not yet emerged from these studies. Neutrophils are crucial regulators of the microenvironment, driving the polarization and proliferation of macrophages. 76 Azurocidin and α-defensins are active agents in the supernatant of stimulated neutrophils, which can induce the polarization of macrophages into a proinflammatory M1 phenotype via β2-integrins on macrophages (Figure 3). Treatment with these proteins results in the release of interferon-γ and tumor necrosis factor from macrophages, which in turn induce macrophage switching into the M1 phenotype. A similar interrelation has been reproduced in a mouse peritonitis model; the phagocytic capacity of the macrophages from neutropenic mice was reduced. However, most of the compromised phagocytic ability was rescued by local application of azurocidin or defensins.77–81 Notably, neutrophil-derived gelatinase-associated lipocalin promotes macrophage polarization toward M2c-phenotype macrophage, which express much more myeloid-epithelial-reproductive tyrosine kinase than M1 or M2a macrophages. 76 Interestingly, the cathelicidin LL37 protein, in contrast to neutrophil granule proteins such as azurocidin and defensin 1–3, exhibits a dual function in regulating the polarizing properties of macrophages. LL-37 induces macrophage anti-inflammatory and resolution responses in acute infections, reducing proinflammatory cytokine secretion without compromising phagocytosis or bacterial clearance.82,83 In contrast, LL-37 activates macrophages in chronic inflammation and increases proinflammatory cytokine release, thus inducing M1 macrophage polarization. 80 In addition, cholesterol crystals in atherosclerotic lesions also counteract neutrophils and promote the formation of NETs, driving the polarization of macrophages into the M1 phenotype and the release of cytokines that activate Th17 cells, resulting in amplified inflammation. 4 Moreover, coculturing M2 macrophages with NETs increases the secretion of proinflammatory cytokines by the M2 macrophages. 84 In vitro experiments with human macrophages differentiated via granulocyte-macrophage colony-stimulating factor have shown M2 macrophage differentiation in coculture with apoptotic neutrophils. 85 However, in vitro experiments with human blood monocyte-derived macrophages have revealed that both apoptotic and viable neutrophils reduced the proinflammatory cytokine release rate and suppress the nuclear factor kappa B signaling pathway in lipopolysaccharide-induced macrophages by inhibiting the upstream TAK1/IKK-β signaling axis. 86

Neutrophils modulate the phenotypic differentiation of macrophages.
Foam cells, also called lipid-laden macrophages, are hallmarks of atherosclerotic initiation. They can be detected only by microscopic examination of a fatty plaque removed from the body. 87 Macrophages internalize modified lipoproteins such as beta, VLDL, acetylated LDL, and Ox-LDL by binding to the scavenger receptors CD36 and CD68 and thus becoming lipid-engorged foam cells. 88 Neutrophil defensin-induced macrophages show increased surface abundance of CD36 and CD38 and contribute to the further enhancement of foam cell formation via LDL receptor-related protein receptors. 89 Since LDL can interact with DNA, 90 it can adhere to NET fibers, which can facilitate LDL aggregation. 91 This DNA–LDL complex triggers foam cell formation via activation of the NLRP3 inflammasome in macrophages by acting as a damage-associated molecular pattern receptor and producing ROS.92,93 In addition, neutrophils can directly phagocytose cholesterol, which may lead to apoptosis or pyroptosis. 94 The clearance of cholesterol-containing debris by macrophages may promote the formation of foam cells.
Neutrophils and plaque destabilization
Atherosclerosis is similar to a volcano that continuously accumulates energy. Lipid cores and intraplaque hemorrhage at the base of the atherosclerotic “volcano”. The crater faces the inner wall of the blood vessel, and the unexploded crater is covered with an uplifted fiber cap, which is covered with an endothelial monolayer. The two sides of the fibrous cap are called plaque shoulders. Neutrophils accumulate in the shoulder regions of atherosclerotic lesions and may even outnumber monocytes/macrophages in these areas. 27 Flow cytometry and confocal microscopy data have shown that neutrophils account for 1.8% of CD45+ leukocytes in advanced plaques in ApoE−/− mice fed a high-fat diet. 27 Furthermore, intravital microscopy has shown that most leukocytes interacting with the endothelium on atherosclerotic lesion shoulders are neutrophils, suggesting the significant recruitment of neutrophils to advanced plaques. 27
ECs protect the fibrous cap, and the stabilization of the fibrous cap depends on the balance of synthesis and catabolism of extracellular matrix components (see Figure 1). Experimental data indicate that neutrophil-derived ROS and proteases promoted superficial erosion in vulnerable plaques.95–97 The damaging effects of ROS released by neutrophils on ECs are described above. 39 The same mechanism may lead to the erosion and destruction ECs outside the fibrous cap in the late stage of atherosclerosis. Here, we focused mainly on disruption of the fibrous cap by proteases released from neutrophils. Myeloperoxidase, an enzyme stored in neutrophil primary granules, is released from neutrophils upon activation by cytokines relevant to atherosclerosis.98,99 When released, myeloperoxidase binds to the extracellular matrix and converts chloride anions and hydrogen peroxide into hypochlorous acid, a potent oxidant and chlorinating species. 100 Therefore, myeloperoxidase is a bridge linking inflammation and oxidative stress with atherosclerosis. 101 Immunohistochemical evidence has demonstrated the presence of myeloperoxidase and hypochlorous acid-modified proteins in atherosclerotic lesions with superficial erosion or plaque rupture.102,103 Further work has revealed that hypochlorous acid associated with sites of inflammation induced apoptosis in ECs. 104 Myeloperoxidase may further promote the survival of neutrophils via ligation of CD11b/CD18 and prolong inflammation. 105 However, Brennan et al. reported that atherosclerotic lesion area is increased in myeloperoxidase-deficient mice; thus, the role of myeloperoxidase in atherosclerosis requires further investigation. 106 In addition, MMP-2 and MMP-9, which are abundant in neutrophil secondary and tertiary granules, play important roles in matrix remodeling and are found in plaque shoulders and regions of foam cell deposition. 107 They are released from activated neutrophils and cleave intact, nonfibrillar and fragmented interstitial collagen, all of which are key components of the subendothelial basement membrane.107,108 However, the relationship between MMP-2 expression and unstable plaques is controversial. Some studies have suggested that MMP2 changes the thickness and thus the stability of the fibrous cap by inducing the migration, proliferation or apoptosis of SMCs.109,110 In the aortic sinus and arch, MMP-2 deficiency resulted in decreased proteolytic activity. It also reduces the migration of SMCs from the medial to the intimal region. A much thicker fibrous cap region that contains SMCs and collagen has been observed in MMP-2+/+:apoE−/− mice compared to MMP-2−/−:apoE−/− mice, suggesting that MMP-2 may modulate plaque stability by inducing SMC movement into the fibrous cap. 111 MMP-2 is also activated by thrombin, increasing local matrix-degrading activity and contributing to cap thinning and weakening.107,112 However, many studies have indicated that MMP2 is unrelated to the stability of plaques.113,114 In contrast to MMP2, MMP9 has been widely recognized for promoting plaque instability. MMP-9 cleaves and increases the chemotactic activity of CXCL1 and CXCL8, thereby amplifying leukocyte tissue infiltration. 115 In addition, neutrophil elastase activates MMP-9, potentiating its activity. 116 Overexpression of auto-activating pro-MMP-9 greatly reduces plaque stability. 117 Similarly, adenovirus-mediated overexpression of MMP-9 disruptes advanced plaques caused by collar implantation. 118
In addition to plaque destabilization via the disruption of the fibrous cap comprising ECs and extracellular matrix, intraplaque hemorrhage promotes the entry route of neutrophils into the plaque. 97 Neutrophils entering plaques in this way release proteases, thereby contributing to plaque destabilization. 97 An additional critical feature of unstable plaques is the necrotic core. Cores become necrotic when apoptotic cells are not cleared by macrophages and subsequently undergo secondary necrosis. As the core continues to grow larger, increasing stress contributes to the fibrous cap. 119 SMCs and macrophages are considered the most frequent sources of necrotic cells in advanced atherosclerotic lesions. However, recent studies have indicated that neutrophils are also important sources of apoptotic and necrotic cells in the necrotic atherosclerotic core. 120
NET formation contributes to plaque instability. The histone H4 derived from intimal NETs forms membrane pores, resulting in SMC necrosis and vascular tissue damage, thereby destabilizing plaques. 96 In addition, NETs can activate the absent in melanoma 2 (AIM2) inflammasome in macrophages, releasing the proatherogenic cytokines IL-1β and IL-18 and leading to unstable atherosclerotic lesions. 121 The use of a genetic variant to increase JAK-STAT signaling via the JAK2V617F mutation imparts a greater risk of premature myocardial infarction, stroke, and clonal hematopoiesis of indeterminate potential and exacerbated atherosclerosis via the AIM2 inflammasome.122,123 Deleting critical inflammasome components, such as caspase 1 and 11, gasdermin D or inhibiting IL-1β reduces necrotic core formation while increasing the stability of plaques. 123
Neutrophils and atherothrombosis
Atherothrombosis is the formation of a thrombus in atherosclerotic arteries, which is the most important feature of advanced atherosclerosis because of its hazardous outcomes, including lethality. In most cases, atherothromboses form after atherosclerotic rupture. Rupture-prone atherosclerotic lesions comprise large necrotic cores and a fibrous cap composed of vascular SMCs and collagen. Activated neutrophils can cause ECs death via apoptosis or produce proteases, which disrupt ECs adhesion to the vessel wall, resulting in endothelial denudation. This endothelial erosion exposes the underlying connective tissue matrix and allows platelets to adhere to the site, resulting in thrombus formation. Therefore, circulating neutrophil counts are strong predictors of acute atherothrombotic events; neutropenia and lymphopenia are also associated with increased cardiovascular risk. 124 Coronary atherothrombotic specimens from patients with acute myocardial infarction contain large numbers of activated neutrophils.125–127 A possible mechanism of neutrophil recruitment to atherothrombotic sites is triggered via complement activation. 128 The sudden rupture of an atherosclerotic plaque triggers activation of thrombin-activated platelets interacting with neutrophils at the site of plaque rupture, resulting in NETs formation and cathelicidin LL37 release, which induce and amplify platelet activation and blood coagulation.129–131 Inhibition of the enzyme peptidyl arginine deiminase 4 (PAD4), which is required for NETs formation, with Cl-amidine has been shown to abrogate NETs formation, attenuate the atherosclerotic burden and arterial thrombosis in mice, and further limit injury in a model of myocardial infarction.127,132 Genetic deficiency of PAD4 decreases acute thrombotic complications of intimal atherosclerotic lesions in mice. 133 In addition, the results from a recent Mendelian randomization study of 547,261 subjects show that greater neutrophil counts are associated with a higher risk of cardiovascular diseases. 134
Therapeutic targeting of neutrophils and future challenges
The above descriptions indicate that neutrophils play essential roles in different stages of atherosclerosis. Therefore, therapeutic strategies for atherosclerosis targeting neutrophils are urgently needed. Despite many failed attempts, ongoing clinical trials are targeting chemokines to inhibit neutrophil infiltration into inflamed tissues; for example, a small-molecule inhibitor of CCR5 is in the pipeline. 135 Neutrophil-derived alarmins, such as S100A8 and S100A9, have been shown to play significant roles in determining the nature and magnitude of the inflammatory response postmyocardial infarction and during the resolution of inflammation. 136 Previous studies have shown that blockade of the S100A8/A9 interaction with its receptors, namely, TLR4 and receptors for advanced glycation end-products, reduces inflammation and disease progression in several inflammatory models and may be repurposed to target inflammation in cardiovascular diseases.137–139 Inhibition of IL-1β release from neutrophils by treatment targeting the NLRP3 inflammasome has been shown to maintain atherosclerosis at a subclinical level. 140 Preventing NET-induced inflammation is another promising strategy for controlling cardiovascular inflammation. NETs chromatin degradation with systemic DNase I treatment has been shown to prevent cardiovascular inflammation and thrombosis. 133 Additionally, the neutralization of histones bound to NETs may be a potential therapeutic approach by reducing monocyte infiltration into atherosclerotic lesions and prevention of SMC death to stabilize atherosclerotic lesions. 64 Inhibiting histone citrullination mediated by the enzyme PAD4 can prevent NETs release and reduce vascular inflammation in mouse models of atherosclerosis. 141 In addition to limiting NETs generation, dissolving NETs is another therapeutic avenue. For example, DNase-1 dissolves NETs by breaking up the DNA strands. DNase has been applied to other processes, such as clearing bronchial mucous enriched with neutrophil products, such as in cystic fibrosis. Experimental and initial clinical studies have shown that infusion of DNase leads to attenuated injury after myocardial infarction caused by ischemia‒reperfusion.25,142 However, potential drawbacks of neutrophil suppression cannot be ignored given the roles of neutrophils in pathogen clearance, tissue repair, mucosal wound healing and inflammation resolution.23,143,144 How to balance the double-edged sword function of neutrophils is a great challenge.
Undoubtedly, more in-depth studies are needed to identify the precise mechanisms by which neutrophils participate in the whole process of atherosclerosis in vivo, such as the mechanism triggering intracellular ROS production in neutrophils and ROS effects on EC injury, apoptosis and necrosis. NETs have been identified at each stage of cardiovascular disease. Nevertheless, whether NETs play different roles at different stages remains unknown. Additionally, exploring whether NETs are involved in crosstalk with SMCs, which are other major sources of foam cells during atherosclerosis, is a challenge. In addition, the effect of neutrophil exosomes on atherosclerosis is an entirely new field, which is not further elaborated herein due to limitations to the review length. Nevertheless, the results of related studies are very instructive and exciting.
Footnotes
Acknowledgements
We thank the Figdraw company for their generosity in providing us with free drawing software.
Author contributions
Xiaojing Zhang: Drafted the manuscript, with critical revision by all other authors. Zhanfang Kang: Literature searching and editing. Dazhong Yin: Writing and editing. Jun Gao: Conceptualization and supervision. All authors have read and agreed to the published version of the manuscript.
Data availability statement
Not applicable.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was financially supported by the Medical Research Fund of Qingyuan People's Hospital (Nos. 20200101, 20190224, 20190227), the 2022 Science and Technology Project of Qingyuan (No. 2022KJJH036), the Medical Research Fund of Guangdong Province (A2021431), and the 2021 and 2022 Research Project of Traditional Chinese Medicine Bureau of Guangdong Province (No. 20211459 and No. 20221470).
Institutional review board statement
Not applicable.
Informed consent statement
Not applicable.