
Abstract
Immune mediated graft loss still represents a major risk to transplant recipients. Creative approaches to immunosuppression that exploit the recipient's own alloregulatory mechanisms could reduce the need for pharmacologic immunosuppression and potentially induce immune tolerance. In the process of studying recipient derived myeloid derived suppressor cells (MDSCs), we identified key alloregulatory MDSC mechanisms, mediated by isolatable proteins IL-4, IL-34, and IL-10. We sought to purify these proteins and fuse them for subsequent infusion into transplant recipients as a means of inducing an alloregulatory response. In this introductory investigation, we leveraged molecular engineering technology to create a fusion protein (FP) of three cytokine coding sequences of IL-4, IL-34, and IL-10 and demonstrated their expressions by Western Blot analysis. Following purification, we tested whether FP IL-4/IL-34/IL-10 (FP1) can protect heart transplant allografts. Injection of FP1 significantly prolonged allogeneic cardiac graft survival in a dose-dependent fashion and the increase of graft survival time exceeded survival attributable to IL-34 alone. In vitro, MDSCs cells were expanded by FP1 treatment. However, FP1 did not directly inhibit T cell proliferation in vitro. In conclusion, newly developed FP1 improves the graft survival in cardiac transplantation mouse model. Significant additional work to optimize FP1 or include other novel proteins could supplement current treatment options for transplant patients.
Introduction
Immunosuppression management remains a challenge for transplant patients and their physicians.1–3 The ideal regimen would theoretically lead to immune tolerance, obviating the need for daily pharmacologic immunosuppression.4,5 Various strategies for immune tolerance have been attempted, with promising results.5,6 Common transplantation tolerance induction protocols have relied on the use of bone marrow transplants to generate mixed-chimerism, but these approaches carry risks of graft-versus-host-disease (GVHD) as well as engraftment syndromes. 7 Rather than bone marrow-dependent strategies, one alternative approach is to leverage the recipient's own immune system to suppress alloreactivity. In this regard, we have focused on myeloid derived suppressor cells (MDSCs) and their ability to suppress anti-donor T cell responses. 8
In our hands, adoptive transfer of recipient derived MDSCs leads to substantial prolongation of heterotopic cardiac graft survival in mice.9–11 However, adoptive transfers are complex therapies that are not easily adaptable to clinical medicine. 8 As such, we hypothesized that a protein which harnessed the T cell suppressive functions of MDSCs, would eliminate the need for cellular infusions, but simultaneously exploit the benefits of MDSCs.
For fusion proteins (FPs) to be optimized for transplantation, protein size, purity, and metabolic turnover must all be addressed. 12 Only a single fusion protein is currently approved for kidney transplantation, Belatacept, which is a mutated form of the receptor CTLA4 (CD152) fused to IgG Fc. 13 Belatacept has shown great promise, but primarily focuses on a classical immunosuppression approach, rather than stimulation of an alloregulatory response.14,15 Fusion proteins that target MDSC expansion have not been developed for the purposes of prolonging allograft survival. 8
IL-34 was recently identified and shares homology with M-CSF by using a common receptor, CSF-1R (CD115) to mediate metabolic pathways and immune response.16–18 Bezie et al. demonstrated that IL-34 serves as a suppressive Treg-specific cytokine. Further, IL-34 greatly expands MDSCs, and subsequently MDSCs expand regulatory T cells. 17 IL-34, like M-CSF, binds the CD115 receptor, driving MDSC expansion.
Interleukin-10 (IL-10) is a well-known T cell suppressive cytokine. 19 Bah et al. showed an association between IL-10 production in T cells and development of immune suppressive MDSCs. Indeed, IL-10 activates S100A9 expression, and S100A9 reprograms myeloid precursors to an MDSC lineage.20,21 IL-10 appears to be crucial for differentiation of MDSCs in vitro.22–25 However, IL-10 and many other cytokines have short half-lives, and are not optimized to expand MDSCs for therapeutic purposes. Beyond IL-10, IL-4 is important for activating as well as maintaining MDSCs. 20 IL-4 also induces MDSCs to express arginase-1, one of the critical mechanisms by which MDSCs suppress T cells, through depleting local levels of L-arginine.26,27
We hypothesized that a fusion protein built from IL-34, IL-10, and IL-4 could act to stimulate an alloregulatory response in a transplant recipient, mimicking the actions of an MDSC adoptive transfer. In this initial exploration of FPs to accelerate MDSC activity to the benefit of transplant recipients, we created a novel multiple cytokine-fused protein IL-4/IL34/IL-10, which we refer to as “FP1”. Building on our prior work and established protocols, we leveraged a heterotopic heart transplant model in MHC disparate mice to determine if FP1 has the potential for immune regulation and graft survival prolongation. We found that FP1 prolonged graft survival by ∼100% in a dose-dependent manner in vivo and promoted BM cell proliferation and increased expansion of MDSCs in vitro. In this article and first description in the literature, we report that a novel multiple cytokine-fused protein, IL-4/IL34/IL-10 is protective to cardiac grafts, and thus it may suggest that further optimization of FP1 or FPs like it might be able to establish immunologic milieus.
Materials and methods
Mice
Wild type C57BL/6J (H-2b) and BALB/c (H-2d) were purchased from Jackson Laboratory (Bar Harbor, ME). Animals were all 6–8 weeks old female. Mice were housed in a specific pathogen-free (SPF) animal facility. The Institutional Animal Care and Use Committees (IACUC) of the University of Maryland Baltimore approved animal protocols. Experiments complied with the Guide for the Care and Use of Laboratory Animals.
Heart transplantation
C57BL/6J hearts were transplanted heterotopically into BALB/c recipients by anastomosing the aorta and pulmonary artery of the graft end-to-side to the aorta and vena cava of recipients. Graft function was monitored every other day by abdominal palpation. Rejection was defined as complete cessation of a palpable beat.
Flow cytometry and antibodies
Cells were treated with anti-mouse CD16/32 (clone 93, eBioscience, San Diego, CA) to block FcR III/II receptors, then stained with antibodies according to manufacturer's protocol and washed two times in FACS buffer (1x PBS with 2 mM EDTA). Brilliant Violet 421 anti-mouse/human CD11b (M1/70), PE-anti-mouse Ly6G/Ly6C (Gr-1) (RB6-8C5), and APC anti-mouse Ly6G (1A8) antibodies were obtained from Biolegend (San Diego, CA) and FITC anti-mouse Ly6C (AL-21) was obtained from BD Pharmingen (San Diego, CA). Stained cells were analyzed using an LSR Fortessa Flow Cytometer (BD Biosciences, San Jose, CA) and results were analyzed with FlowJo software (Tree Star, Ashland, OR).
Cell proliferation assay
To detect the proliferation of T cells, T cells were enriched by magnetic bead negative selection (Stemcell Technologies, Vancouver, Canada) from WT BALB/c mice, and labeled with 5 µM Carboxy fluorescein succinimidyl ester (CFSE) (Thermo Fisher), in 1 ml of PBS for 15 min at 37°C. A total of 1 × 105 CFSE-labeled T cells were plated in complete media (RPMI 1640, 10% FBS, 20 units/ml penicillin, 50 mg/ml streptomycin) supplemented with 2 mmol/l l-glutamine and 0.05 nmol/l 2-mercaptoethanol onto flat-bottomed 96-well plates (Corning, B.V.) coated with 5 µg/ml anti-CD3 and 2 µg/ml anti-CD28 (eBioscience). IL-4/IL-34/IL-10 fusion protein was added at various concentration (0. 1. 5. 10. 25. 50 µg/ml). Following incubation for 3 days at 37°C in a 5% CO2 incubator, cell proliferation was assessed flow cytometric analysis of CFSE dilution.
Bone marrow cell culture
For analysis of in vitro expanded MDSCs, bone marrow cells were acquired from tibias and femurs of BALB/c as described previously. 28 Tibias and femurs were removed using sterile techniques, and bone marrow was flushed with PBS. Red blood cells were lysed with ammonium chloride. Total bone marrow cells were planted into 100-mm dishes (Corning, USA) and further cultured in RPMI 1640 culture media supplemented with 10% FBS, 100 µg/ml streptomycin and 100 U/ml penicillin, 10 mM HEPES, 2 mM L-glutamine, 1 mM pyruvate, 0.02 mM 2-ME in the presence of either 20 ng/ml GM-CSF (R&D system, Minneapolis, MN) with or without 20 ng/ml of IL-4/IL-34/IL-10 fusion protein. During incubation, fresh complete medium was replaced every other day. Following 6day incubation, CD11b + Gr-1+ cells (total MDSCs), CD11b + Ly6G + Ly6Clo (PMN-MDSCs), and CD11b + Ly6G−Ly6Chi (M-MDSCs) were analyzed, and total cell number was counted.
Statistical analysis
Data are presented as mean value ± standard error (SEM). The Student's t-test was used to compare the difference of mean between each of two groups. One-way ANOVA was used to compare significant difference between more than two groups. The survival curves were analyzed using Log rank (Mantel-Cox) test. A p value of <0.05 was considered significantly different. Data were analyzed using Graph Pad Prism V9.0 (LaJolla, CA, USA).
Engineering, expression, and purification of the Il-4/Il-34/Il-10 fusion protein FP1
The gene corresponding to human IL-4/IL-34/IL-10 fusion protein was codon optimized for the expression in Escherichia coli and cloned into pET28 expression vector without any affinity tags. The construct was transformed into BL21(DE3) competent cells. Large scale cultures were grown at 37°C until OD reached 0.6, protein expression was induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) at 37°C. After 3 h, cell cultures were harvested by centrifugation at 4000 r.p.m. for 15 min at 4°C and collected cell pellets were resuspended in lysis buffer (50 mM Tris/HCl pH 8.0, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), and 0.1 mM phenylmethylsulfonyl fluoride (PMSF)); lysed by sonication using 50% power for 2 min (5″ on/off), and centrifuged at 25,000 g for 45 min at 4°C to remove cell debris and insoluble proteins. The pellets containing inclusion bodies of human IL-4/IL-34/IL-10 fusion protein were washed 3× times with lysis buffer supplemented with 1% Triton X-100 followed by resuspension in lysis buffer supplemented with denaturing buffer 5 M guanidine HCl pH 8.0, 2 mM reduced/0.2 mM oxidized glutathione (GSSG/GSH) and incubation overnight with mild stirring. Protein concentration of solubilized mix of proteins was adjusted to 2.5 mg/ml using denaturing buffer. The refolding process started with a dialysis against 50 mM Tris-HCl pH 8.0, 300 mM NaCl, 1 mM EDTA, and 0.5 ml ProteCEASE-50AAnion protease inhibitor cocktail for 24 h. The dialysis membrane was changed to a final buffer-exchange against buffer A (20 mM Tris-HCl pH 9) for 24 h to prepare for HiTrap™ Q FF anion exchange column previously equilibrated with buffer A. Human IL-4/IL-34/IL-10 fusion protein, FP1, was eluted between 280 mM and 350 mM of NaCl gradient. Fractions were identified by a single band via SDS-PAGE gel at the correct molecular weight for FP1 (70 kDa). The identity of this band was confirmed via western blot (WB) using human IL-10 monoclonal mouse IgG1 Clone#127107 (R&D Systems; MAB2172-100) antibody, which detects for hIL-10 and hIL-34 monoclonal mouse IgG1 Clone # 578416 (R&D Systems; MAB5265) antibody, which detects for hIL-34. Additional confirmation of the molecular weight was provided for the human IL-4/IL-34/IL-10 fusion protein upon the final purification step by size exclusion chromatography, in which FP1 eluted from a HiLoad®16/600 Superdex®200 previously equilibrated with endotoxin-free Dulbecco's phosphate-buffered saline buffer (pH 7.2) in a fraction consistent with its molecular weight, as compared to a calibration curve of molecular weight standards including: bovine thyroglobulin (670 kDa, 56.1 ml), bovine γ-globulin (158 kDa, 72.9 ml), chicken ovalbumin (44 kDa, 87.0 ml), and equine myoglobin (17 kDa, 97.7 ml) indicated with arrows (Bio-Rad # 1511901). Purity and quality of purified protein was assessed of pure FP1 (>99%) by SDS-PAGE, western blot analyses, and native-PAGE. No endotoxin was detected using this purification protocol as monitored quantitatively with the ToxinSensor ™ chromogenic LAL endotoxin assay kit (UV absorbance at 545 nm). Purified FP1 was filtered under sterile conditions in endotoxin free buffer, aliquoted (100 μL), and frozen at −80°C for later use in cell and in vivo studies such that no more than one freeze-thaw cycle was used for any experiments. Protein QC was measured with ELISA kits from OriGene: Human IL-10 ELISA Kit, 1 × 48-well (pre-coated) CAT#: EA102154
Results
Generation of Il-4/Il-34/Il-10 fusion protein, FP1
Recombinant human IL-4/IL-34/IL-10 fusion protein (FP1) was highly expressed in large E. coli cell cultures into inclusion bodies for the first time. The use of high temperature during protein expression (37°), FP1 induction of expression at a relatively high cell density (A600nm = 0.8–0.9), and the use of a strong promoter system resulted in the FP1 fusion going to inclusion bodies. FP1 was recovered using low detergent concentration and chaotropic agents followed by washing with low concentrations of detergents like Triton X-100 to obtained highly purified and endotoxin free inclusion bodies that lacked membrane fragments. Further purification and refolding protocols provided active human FP1 (Figure 1), which was accomplished specifically by anion exchange and size exclusion chromatography steps. Analyses of the molecular weight of FP1 was fully consistent with the IL-4/IL-34/IL-10 protein gene product, which eluted as a monomer with an apparent mass of ∼ 70 kDa, as determined by sizing studies completed during the gel permeation chromatography step. The ELISA assays were run to detect the protein. IL4 and IL10 were detected in a dose-dependent manner, but IL34 was not.

Generation of IL-4/IL-34/IL-10 fusion protein FP1.
FP1 significantly prolongs cardiac allograft survival
To test the functional immune effects of FP1, we used our established mouse cardiac transplant model.11,29 We treated H-2d BALB/c recipients of fully MHC-disparate H-2b C57BL/6 (B6) heart allografts with either phosphate buffer saline (PBS, group CON) or FP1 (5 mg/Kg weight i.p.) as either a single dose on day 0 (group FP1) or three doses on days 0, 3, and 6 (group FP3), post-transplantation (Figure 2A) and monitored heart allograft survival (Figure 2B). Treatment with FP1 among BALB/c recipients prolonged BALB allograft survival from a mean survival of 6 days (without FP) to 10.5 days (with 1 dose of FP, FP1) days (p < 0.005) (increase in survival of 75%). Multiple doses of FP1 to recipients more increased grafts survival to mean graft survival of 22 days (with 3 doses of FP, FP3) (p < 0.05), more than tripling the survival times of untreated or IL-34 alone – treated animals (Supplemental Figure 1). Graft survivals of all the animals transplanted showed in Table 1. These data suggest, that while insufficient for tolerance induction, FP1 was immuno-protective, and prolonged graft survival in mouse recipients of MHC disparate heart transplants.

IL-4/IL-34/IL-10 fusion protein prolonged cardiac allograft survival. (A) 5 mg/Kg of Il-4/IL-34/IL10 fusion protein was intraperitonially injected on day 0 for single dose group (FP1) or on day 0, 3, and 6 for multiple dose group (FP3) post-transplantation. (B) Graft survival was analyzed by abdominal palpation. Survival curves show significant difference between control (No treatment) and FP-treated groups (FP1 or FP3) by log-rank (Mantel-Cox) test, *p < 0.05, Mean survival time (MST, days).
Graft survivals of each case.

FP1 does not suppress T cell proliferation in vitro
Given the recent demonstration 30 that IL-34 can promote the proliferation of Th17 cells in a dose-dependent manner, we first sought to determine the effect of FP1 on the proliferation of T cells in vitro. We used flow cytometry to detect proliferation of T cells. T cells were enriched from a single cell pool derived from spleen and lymph nodes procured from WT BALB/c mice and were then incubated in the presence of different concentrations of FP1 (0, 1, 5, 10, 25, and 50 ug/ml). Following 3 days of incubation, CD4 + T cells or CD8 + T cells were collected, washed, and counted. Gating for T lymphocytes (Figure 3A), proliferation was assessed by detection using CFSE expression (Figure 3B). The frequency of proliferating cells stimulated with CD3/CD28 antibodies were 48.7 ± 0.117%, and 62.4 ± 0.186% among CD4+ T cells and CD8 + cells, respectively. FP1 did not change the proliferation capacity of CD4 T cells nor CD8 T cells in the range of different concentrations (0, 1, 5, 10, 25, and 50 ug/ml) used in this experiment (Figure 3C).
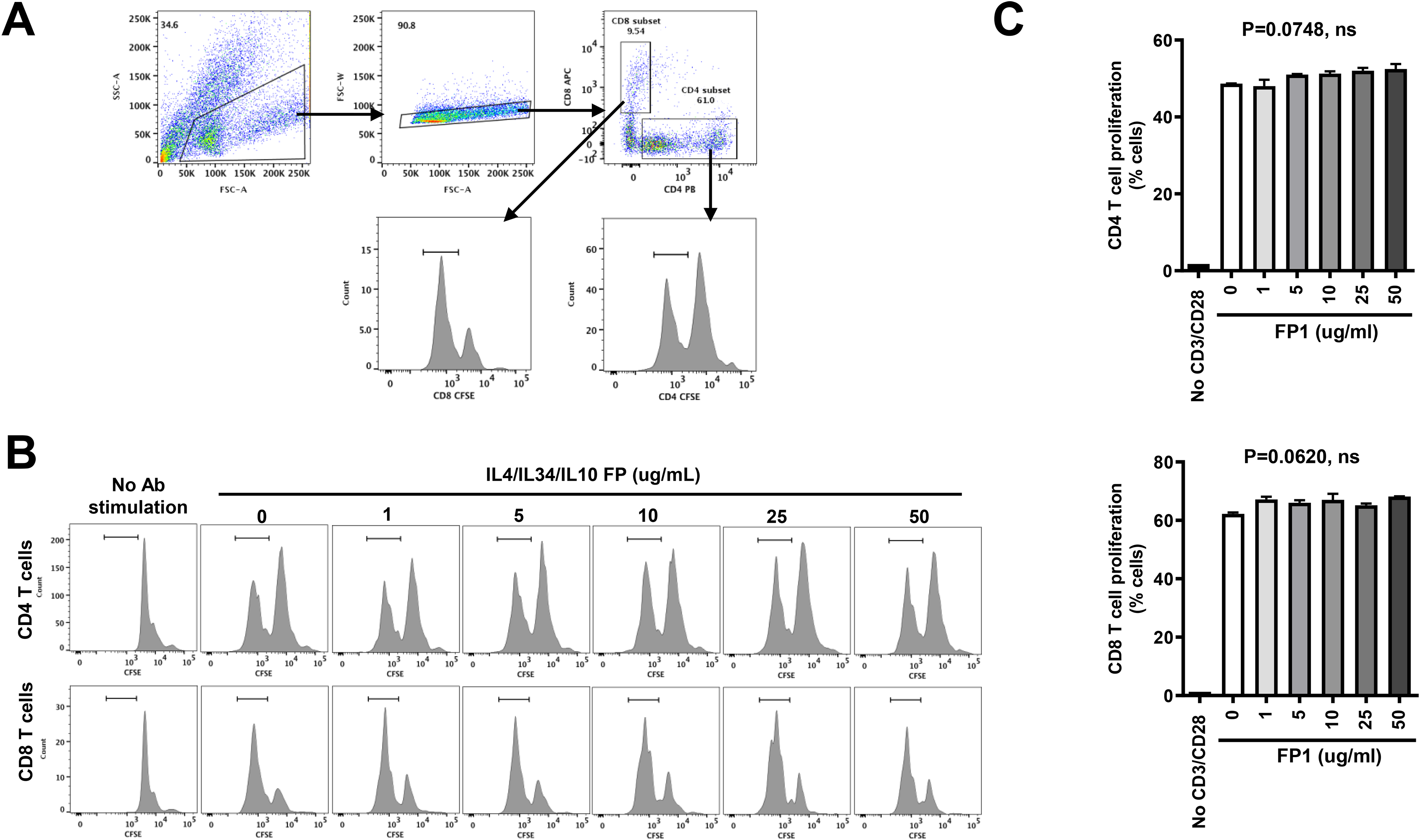
IL-4/IL-34/IL-10 fusion protein does not alter proliferation capacity of T cells. T cells were obtained and cultured as mentioned in the section of Materials & Methods in the presence/absence of IL-4/IL-34/IL-10 fusion protein (20 ng/ml). Following incubation for 3 days, cells were collected and analyzed for proliferation by flow cytometry of diluted CFSE. (A) The gating strategy for proliferated CD8+/CD4 + cells. (B) Histogram plots show representative flow cytometric data of proliferating cells in CD4 + or CD8 + cells. (C) Bar charts show the frequency of proliferated cells of CD4 + or CD8 + cell. The differences between seven groups were tested by ANOVA and after post Tukey test. T cells were stimulated with CD3 & CD28 antibodies during incubation for 3 days before flow cytometry assay.
FP1 promotes expansion of CD11b + Gr-1+ MDSCs in vitro
IL-34 was included in FP1 to stimulate MDSC expansion. We tested the effect of FP1 on the proliferation and differentiation of BM cells in vitro. We isolated bone marrow cells from WT BALB/c mice and examined the proliferation capacity of the cells and the frequency of CD11b + Gr-1+ MDSCs in the presence and absence of FP1 (20 ng/ml). Following 6 days of incubation, the addition of FP1 the increased proliferation of BM cells 1.7-fold when compared to the control without FP1 (FP1 vs control; 2.2 × 106 vs 1.3 × 106 cells, ***p < 0.001) (Figure 4A). Notably, the frequency of CD11b + Gr-1 + cells (total MDSCs) among the cell population was increased when cells were cultured in the presence of FP1 (FP1 vs control; 51.9% vs 40.1%, *p < 0.05) (Figure 4B) with significant increase in absolute cell numbers (FP1 vs control; 1.1 × 106 vs 0.5 × 106 cells, data not shown). In addition, the frequency of PMN-MDSC and M-MDSCs, two major subsets of MDSCs, were also increased by FP1 from 25% to 33% (Figure 4C, *p < 0.05) and from 22% to 28% (Figure 4D, ns), respectively. Our observation of expansion of MDSCs, a potent immune suppressor, suggests FP1 promotes expansion of MDSCs, potentially to the benefit of transplant recipients.

IL-4/IL-34/IL-10 fusion protein promotes in vitro MDSCs expansion. Bone marrow cells were obtained cultured as mentioned in the section of Materials & Methods in the presence/absence of IL-4/IL-34/IL-10 fusion protein (20 ng/ml). Following incubation for 6 days, cells were collected and analyzed for total MDSCs, PMN-MDSCs, and M-MDSCs. (A) The representative histogram images and quantitative analysis of percentage of MDSC subsets induced 6 days after incubation. Data were analyzed as mean value ± SD and student t-test was used to assess the result significance. *p < 0.05, ***p < 0.001, compared with the control group, ns, not significant.
Discussion
Here we present an early, and exciting approach to alloregulation, but further work is clearly required. Creative approaches to immunosuppression protocols and tolerance induction are needed in order to innovate classical strategies of pharmacologic immunosuppression which carry risks of infection, malignancy, heart disease, and non-compliance.31–33 MDSCs are an attractive solution for modifying a recipient's immune response after transplant, and possibly tolerance induction. However, the administration of MDSCs via adoptive transfer is complex, likely very expensive, and carries risks of off-target side effects because MDSCs have many functions that extend beyond alloregulation.8,29 To that end, an optimal MDSC-based solution would be one which provided transplant patients the benefits of Myeloid Derived Suppress Cell infusions, but without the “cell.”
Our prior work showed that MDSC adoptive transfers plus a single dose of anti-CD40L was highly effective in prolonging heterotopic cardiac allograft survival, and potentially achieving tolerance.9–11 However, when anti-Gr1, an antibody to MDSCs, was administered to recipients of this protocol, animals rejected their allografts within days. This suggested a direct, MDSC-based alloregulatory effect of the adoptive transfer. Importantly, the abrogation of prolonged survivals using anti-Gr1 was time dependent. For example, if anti-Gr1 was given more than 3 weeks after the adoptive transfer, treatment with anti-Gr1 failed to provoke rejection. This suggested that perhaps MDSCs, which were allowed to expand under the cover of T cell suppression with anti-CD40L, stimulated a secondary peripheral regulatory response in the recipients which was providing ongoing alloregulation and protecting the cardiac allograft. 9 Indeed, further studies showed that Tregs were increased in animals receiving MDSC adoptive transfers, 2–3 weeks after treatment.9,34,35
With these data in mind, and knowing that adoptive transfers are not clinically appealing, we sought to mimic the combination of 1) T cell suppression, 2) MDSC expansion, and 3) MDSC activation/suppression by using key proteins which could be fused together. If effective, this strategy would be highly clinically relevant, and potentially provide a pathway to immune tolerance by leveraging the T cell suppressive effects of recipient derived MDSCs. To this end, we selected the fusion of IL-34 for MDSC expansion, IL-10 for T cell suppression, and IL-4 for MDSC activation and suppression.
Here, we describe our first experience and observations of this longitudinal approach to innovating immunosuppression. Though an early study, we found that FP1, was soluble, purifiable, and appropriate for treatment in animals. From a protein structure standpoint, we found that FP1 was monomeric in solution via gel permeation chromatography with a molecular weight, 70 kDa, as expected for a FP1 monomer. Likewise, the refolding procedures used to provide active cytokine epitopes were identified by western blot analyses. Further indication that FP1 was efficacious was indicated was by several in vitro and in vivo studies. These include with the observation that FP1 promoted MDSC expansion, consistent with an active IL-34 component. Further, in vivo, FP1 was safe, and animals tolerated treatment. FP1 prolonged graft survival in recipients of heterotopic cardiac transplants as well.
FP1 was not effective in vitro at suppressing T cells. The reason for this lack of T cell suppression is not yet clear. Firstly, it is possible that the geometry of IL-10 within FP1 precluded IL-10's binding to IL-10R. This could be remedied by structurally altering FP1 in future experiments. 36 Separately, IL-10 is known to have a short half-life, and IL-10 would need to be present for days (and potentially weeks) to get a suppress alloreactive T cells long enough for MDSCs to sufficiently expand and generate ongoing alloregulatory responses including the generation and expansion of Tregs. 37
We freely acknowledge that this is only the initial exploration of FP1 which was designed to align the effects of early T cell suppression and prolonged alloregulation driven by a recipient's MDSC response. Inclusive of additional controls, new experiments, and modifications to FP1 structure, there are shortcomings of this initial work, and future effort is required to comprehensively understand all of FP1's immune effects. IL-4 and IL-10 were not included as specific controls because IL-4 alone would not be expected to prolong survival and because IL-10 is only immunosuppressive and is not likely to instigate a tolerogenic response. 38 Further, many prior studies have previously shown that lL-10 controls T cell proliferation.39,40 However, future work to optimize FP1 will require in depth analysis of how to modify the contributions of IL-4 and IL-10 to FP1. For example, FP1 will be compared with IL-10 directly to determine 1) does IL-10 need to be repositioned or 2) replaced by a different immunosuppressive cytokine such as IL-35. 41
The graft survival of animals treated by FP1 and FP3 were twice and triple, respectively, compared to those non-treated with FP1. However, the survival is still quite short when we compare it to the resultsthat we achieved with MDSC adoptive transfers. 9 This may be from lack of T cell suppression (IL-10, as suggested above), or from limited MDSC expansion by IL-34. IL-34 is not the only MDSC expanding agent, and it is possible that we could consider the use of CSF1, or alternative ligands for CD115. On the other hand, we do not yet know the half-life of FP1, and it may be that simply prolonging the half-life allows better T cell suppression and MDSC expansion. To address these issues, we will extend the half-life of FP1 first via protein engineering methods, which improves the stability and efficacy of FP1, but simultaneously taking care not to induce any off-target immunoreactivity.
In conclusion, we report on the first experience with FP1, a novel fusion protein designed to leverage the potential benefits of MDSC responses in transplant recipients. While there are limitations to the function of FP1, and while we do not yet understand comprehensively how FP1 impacts immunity in recipients, FP1 suggests there may be value in development of fusion proteins which seek to instigate sustainable alloregulatory effects after transplantation that could minimize or obviate the need for ongoing pharmacologic immunosuppression.
Footnotes
Data sharing statement
The large majority of data generated or analyzed during this study are included in this published article [and its supplementary information files]. However, datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health, (grant number NIH R21AI166272).
Supplemental material
Supplemental material for this article is available online.