
Abstract
Allergic rhinitis (AR) is a nasal mucosal inflammatory disease mediated by environmental allergens. At present, the relationship between the IL-33/ST2 axis, ERK1/2 pathway and AR progression needs further exploration. In our study, an AR model was constructed in vitro by treating HNEpC cells with Der p1. qRT-PCR was applied to assess the mRNA levels of IL-33, ST2, TNF-α, IL-6, and IL-8. Western blotting was used to measure the protein levels of IL-33, ST2, and the downstream proteins p-ERK1/2, ERK1/2, p-RSK, and RSK. IL-6, IL-8, IL-33, and TNF-α protein levels in cell supernatants were evaluated by ELISA. Flow cytometry was performed to check cell apoptosis of HNEpC in the presence or absence of Der p1. Our results indicate that the relative levels of IL-33, ST2, TNF-α, IL-6, and IL-8 were increased significantly in the AR model group. The above effects were notably reversed after transfection with shIL-33 or shST2. IL-33 stimulation further resulted in the increase in both ST2 and inflammation-associated cytokines, and these effects were restored after shST2 treatment. Also, the levels of inflammatory factors induced by IL-33 stimulation or ST2 overexpression were reversed after applying an ERK1/2 pathway blocker. In conclusion, IL-33/ST2 mediated inflammation of nasal mucosal epithelial cells by inducing the ERK1/2 pathway.
Introduction
Allergic rhinitis (AR) is a common nasal mucosal inflammatory disease mediated by environmental allergens through IgE. Its clinical manifestations are nasal itching, sneezing, and nasal congestion, which can induce asthma. 1 In recent decades, the incidence of AR has not improved significantly, and still maintains a high level. Treatment of AR can prevent and control the occurrence of asthma, so it has currently become one of the most important diseases in the respiratory system. 2 Current research suggests that the pathological process of AR is complex, and the disease is known to be caused mainly by exposure of susceptible populations to various potential allergens. However, the mechanism of occurrence and development of AR is not clear.
IL-33 is an IL-1 family cytokine newly discovered in 2005, and it functions by binding specifically to its receptor suppression of tumorigenicity 2 (ST2), forming the IL-33/ST2 axis.3,4 The IL-33/ST2 signal transduction pathway is related closely to allergic diseases, autoimmune diseases, and cardiovascular diseases.5,6 IL-33/ST2 can not only promote the accumulation of eosinophils, basophils, mast cells, and other inflammatory cells in the nasal mucosa, but also promotes the release of cytokines, thereby mediating the development of allergic diseases.7,8 Chronic pulmonary obstructive disease (COPD), is a progressive inflammatory condition, in which an increased expression of IL-33 and the ST2 receptor, as also seen in asthma, has been observed. 9 Studies have shown that IL-33 level in serum of AR patients is increased, and IL-33 and ST2 expression levels are also increased in nasal epithelial cells, suggesting that IL-33/ST2 might play an important role in AR. 10 Therefore, the relationship between the IL-33/ST2 axis and AR progression needs further exploration.
ERK1/2 is a member of the MAPK family. 11 ERK1/2 is an important signal transduction system in cells, which plays an important role in various cell functions such as growth, development, division, differentiation, and apoptosis. 12 ERK signaling activation can promote the expression of a variety of inflammatory cytokines, including TNF-α, IL-6, IL-8, and others.13–15 Studies have shown that IL-33 promotes the secretion of IL-6 via the ST2–ERK1/2 pathway in gastric cancer cells. 16 However, the molecular mechanisms of ERK1/2 in AR still need to be explored.
Based on previous studies, we hypothesized that IL-33/ST2 mediates inflammation of nasal mucosal epithelial cells via regulating ERK1/2 signaling. In this study, the role of the IL-33/ST2/ERK1/2 axis in the inflammatory response of nasal epithelial cells was detected and analyzed by establishing a model of nasal mucosal epithelial cell sensitization. Thereby, the relationship between IL-33/ST2 and ERK1/2 and AR can be further clarified, which provides a more experimental basis for the prevention, diagnosis, and treatment of AR in clinical work.
Materials and methods
AR model establishment
The human nasal mucosal epithelial cell line HNEpC was obtained from ATCC (Manassas, VA, USA). HNEpC cells were cultured in DMEM (Invitrogen, Gaithersburg, MD, USA) with 10% FBS (Invitrogen). To establish the AR model, cells were treated with Der p1 (10 mg/ml) for 24 h. For IL-33 stimulation, cells were treated with 50 ng/ml human recombinant protein IL-33 (Thermo Fisher Scientific, Waltham, MA, USA). To block ERK1/2 signaling, HNEpC cells were treated with BVD-523 (5 μM, MedChem Express, Monmouth Junction, NJ, USA).
Plasmid construction and cell transfection
For IL-33 knockdown, shRNA IL-33 (shIL-33, Biomics, Nantong, China) was transfected into HNEpC cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. To overexpress ST2 (OE-ST2) or block ST2, the overexpression plasmid pcDNA3.1-ST2 or shST2 (Biomics) was transfected into HNEpC cells using Lipofectamine 2000 (Invitrogen).
Quantitative real time-PCR
Trizol (Thermo Fisher Scientific) and PrimeScript RT reagent Kit with gDNA Eraser (Takara, Dalian, China) were used for total RNA extraction and reverse transcription, respectively. Quantitative real time-PCR (qRT-PCR was applied using a SYBR Green qPCR Master Mix Kit (Takara) following the protocol of the manufacturer. Primers for IL-33, IL-8, IL-6, TNF-α and ST2 were synthesized by Sangon Biotech (Shanghai, China) and are listed in Table 1. The amplification condition were 95°C, 10 min, followed by 40 cycles each at 95°C, 15 s, 60°C, 30s, and 72°C, 30s. The relative expression levels were calculated using the 2–ΔΔCq method. GADPH was selected as internal reference gene.
Primers for quantitative real-time PCR.

Extraction of total proteins and Western blotting assay
RIPA buffer (Thermo Fisher Scientific) and BCA protein assay Kit (Beyotime) were applied for total protein extraction and concentration detection, respectively. Proteins were transferred to PVDF membranes (Sigma-Aldrich, Saint Louis, MO, USA) after 10% SDS-PAGE separation. The PVDF membranes were blocked with skimmed milk (5%) for 1 h, followed by incubation with primary Abs (all from Cell Signaling Technology, Danvers, MA, USA): anti-IL-33 (1:2000), anti-ST2 (1:2000), anti-p-RSK (1:2000), anti-RSK (1:1000), anti-p-ERK1/2 (1:2000) and anti-ERK1/2 (1:2000), at 4°C overnight. Membranes were then washed with PBS buffer, and incubated with biotinylated secondary Abs (1:10,000, Cell Signaling Technology). Protein expression levels were measured by an enhanced chemiluminescence detection Kit (Thermo Fisher Scientific). Proteins were quantified using Quantity One software (Bio-Rad Laboratories, Hercules, CA, USA).
ELISA
ELISA kits for measurement of TNF-α, IL-6, IL-8 and IL-33 were obtained from Abcam (Cambridge, MA, USA). ELISA plates were coated with mAbs then incubated at 4°C overnight. PBS containing 10% FBS (Sigma-Aldrich) was added to the plates and blocked for 1 h. The samples were diluted, added to the ELISA plates, and incubated at room temperature for 2 h. Biotin-labeled Ab (1:80 diluted) and avidin-labeled HRP were added to the plates, which were then incubated for 1 h. After adding chromogenic reagent and incubating in the dark for 30 min, the plates were read at 450 nm immediately using a THERMO FISHER Multiskan FC device (Thermo Fisher Scientific).
Measurement of apoptosis
The apoptosis assay was performed using an Annexin V-FITC Apoptosis Detection Kit according to the manufacturer’s protocol (Sigma-Aldrich). In brief, cells were seeded at a density of 1.0 × 105 cells/well in a six-well culture plate and treated with Der p1 for 24 h. The cells were then harvested with 0.25% trypsin, washed twice with cold PBS, and suspended in 500 μl binding buffer supplied by the manufacturer. The cells were then incubated with 5 μl Annexin V and 5 μl propidium iodide reagent in the dark for 20 min. Analysis was performed using a BD FACS Aria flow cytometer (Becton Dickinson, San Jose, CA, USA). At least 10,000 cells were analyzed in each treatment. 17
Statistical analysis
SPSS software (Version 22.0, SPSS, Chicago, IL, USA) was used for statistical analysis. All experiments were repeated at least three times, and one representative result was presented. All data were presented as mean ± SD. Student’s t test was conducted for two group comparison. One-way ANOVA followed by Tukey’s post hoc test was carried out to compare three or more groups. P < 0.05 represented significant difference.
Results
Sensitization of nasal mucosal epithelial cells leads to activation of IL-33/ST2 signaling to promote inflammation
To investigate the relationship between sensitization of nasal mucosal epithelial cells and inflammation, HNEpC cells of an AR model were constructed by Der p1 (10 mg/ml) stimulation for 24 h. The results of qRT-PCR showed that the relative mRNA levels of IL-33, ST2, TNF-α, IL-6, and IL-8 were significantly increased in the AR group compared with the control group (Figure 1a). Subsequently, the protein levels of IL-33 and ST2 were measured by Western blotting assay. As shown in Figure 1b, the protein expression levels of IL-33 and ST2 were up-regulated after cells were sensitized (AR group), indicating that sensitization induced IL-33/ST2 activation. The results of ELISA were consistent with the above results, indicating that the expression levels of IL-33, TNF-α, IL-6, and IL-8 were markedly increased in AR-cells compared with those in control group (Figure 1c,d). The apoptotic rate of AR cells was also higher than that of the control group (Figure 1e). Collectively, these results suggest that sensitization of nasal mucosal epithelial cells led to activation of the IL-33/ST2 axis to promote an inflammation response.
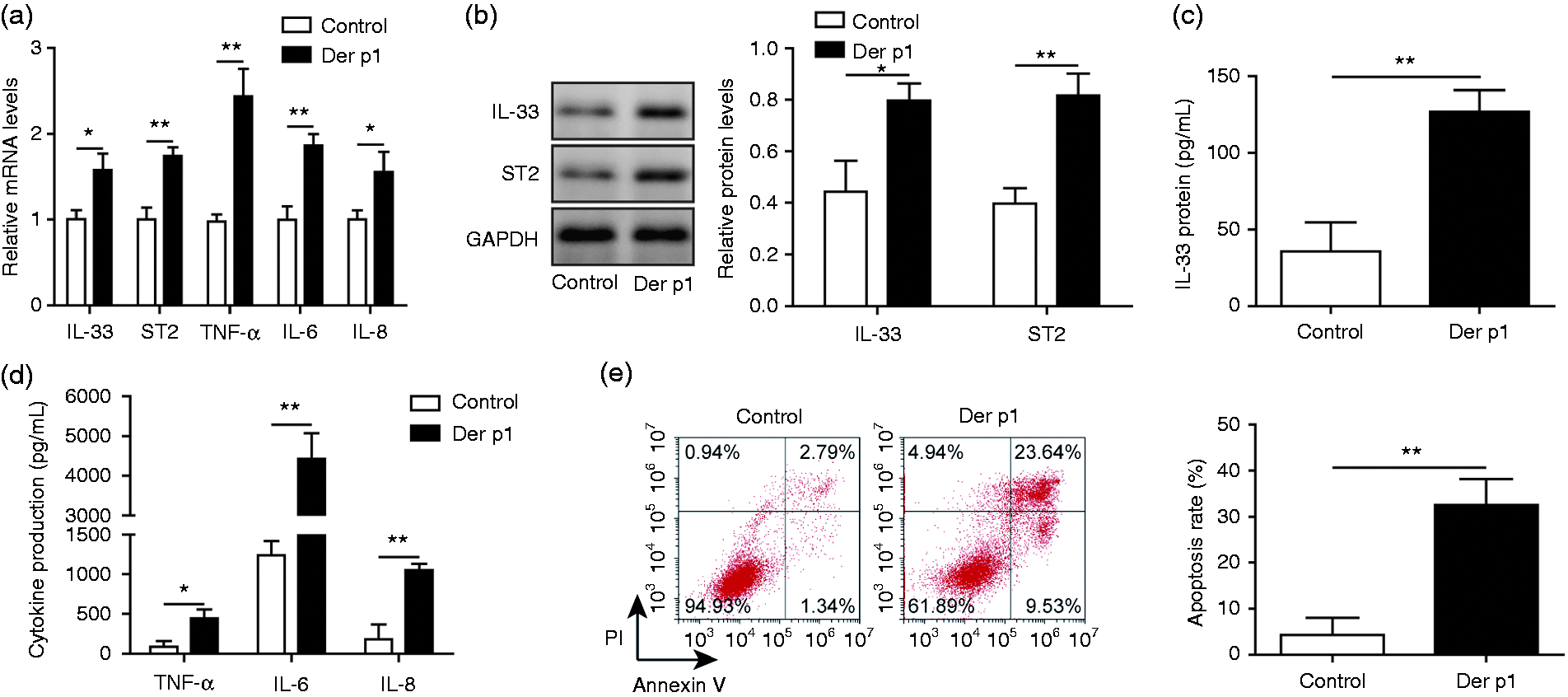
Sensitization of nasal mucosal epithelial cells leads to activation of IL-33/ST2 signaling to promote inflammation. (a) qRT-PCR was performed to analyze the relative mRNA levels of IL-33, ST2, TNF-α, IL-6, and IL-8. (b) Western blotting was used to analyze the protein levels of IL-33 and ST2. (c) ELISA was applied to assess the protein level of IL-33. (d) ELISA was applied to assess the levels of TNF-α, IL-6, and IL-8. E, Flow cytometry was used to check cell apoptosis of HNEpC cells. AR model cells were constructed by treatment with 10 mg/ml Der p1 for 24 h. Cells were divided into two groups: control group and Der p1-treated group. *P < 0.05 and **P < 0.01.
Nasal mucosal epithelial cells mediate cellular inflammation through IL-33/ST2 signaling
To explore and verify whether IL-33/ST2 signaling is involved in mediating the inflammatory response of nasal mucosal epithelial cells, cells of the AR model were transfected with shIL-33 or treated with shST2. The relative mRNA expression levels of ST2 were notably decreased after transfection with shIL-33 or shST2, but shST2 transfection showed no effects on the expression level of IL-33 (Figure 2a). The results of Western blotting experiment showed that protein levels of IL-33 and ST2 were reduced after transfection with shIL-33 in cells of the AR model (Figure 2b). The trend of shST2 treatment showed the little effect on IL-33 expression, and significantly inhibited ST2 expression (Figure 2b). Next, we verified the effect of transfection with shIL-33 or shST2 on AR cells by ELISA and qRT-PCR. As expected, the expression levels of TNF-α, IL-6, and IL-8 were down-regulated after AR cells were transfected with shIL-33 or shST2 (Figure 2c,d). Collectively, these data confirmed and verified that nasal mucosal epithelial cells mediate cellular inflammation through IL-33/ST2 signaling.
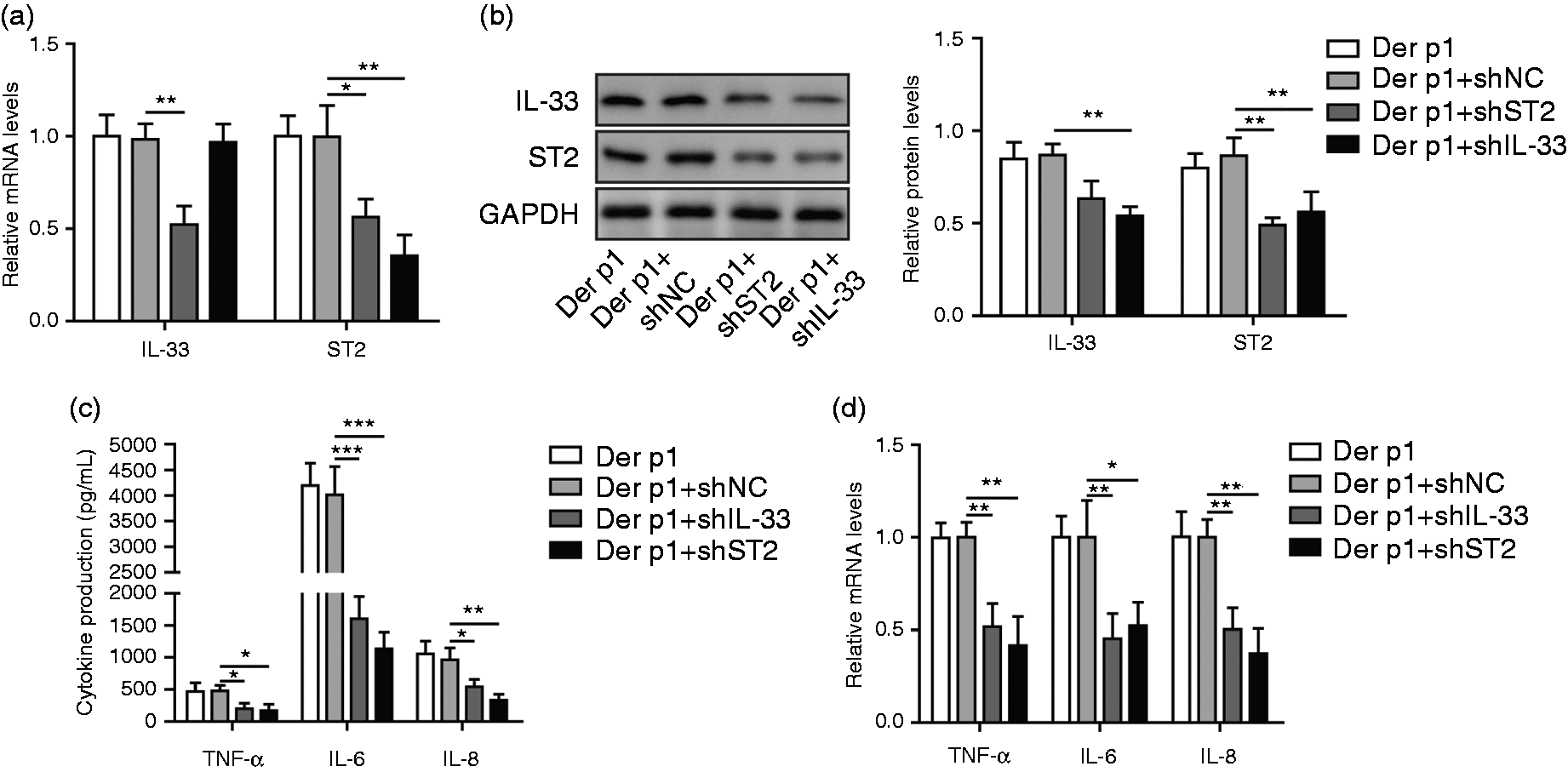
Nasal mucosal epithelial cells mediate cellular inflammation through IL-33/ST2 signaling. (a) qRT-PCR was performed for IL-33 and ST2 analysis. (b) Western blotting was used for IL-33 and ST2 levels analysis. (c) ELISA was used for inflammation-associated cytokine levels analysis. (d) qRT-PCR was performed for TNF-α, IL-6, and IL-8 levels analysis. AR cells were divided into four groups: Der P1-treated group, Der p1+shNC group, Der p1+shIL-33 group, Der p1+shST2 group. *P < 0.05, **P < 0.01, and ***P < 0.001.
IL-33 mediates inflammation of nasal mucosal epithelial cells through ST2 receptor
To investigate the effect of ST2 receptor on the IL-33-mediated inflammatory process in nasal mucosal epithelial cells, cells of the AR model were stimulated with IL-33 and transfected with shST2 vector. qRT-PCR data illustrate that IL-33 treatment resulted in an increase in both ST2 and inflammation-associated cytokines (Figure 3a). However, these effects were restored after shST2 treatment (Figure 3a). As assessed by Western blotting assay, the ST2 protein level was significantly up-regulated in AR cells treated with IL-33, and this effect was reversed by shST2 treatment (Figure 3b). Furthermore, ELISA assay proved that inflammation-associated cytokines were up-regulated after IL-33 stimulation, and the changes were reversed by shST2 treatment, with the same change seen in mRNA levels (Figure 3c). Therefore, we identified that IL-33 mediates inflammation of nasal mucosal epithelial cells through the ST2 receptor.
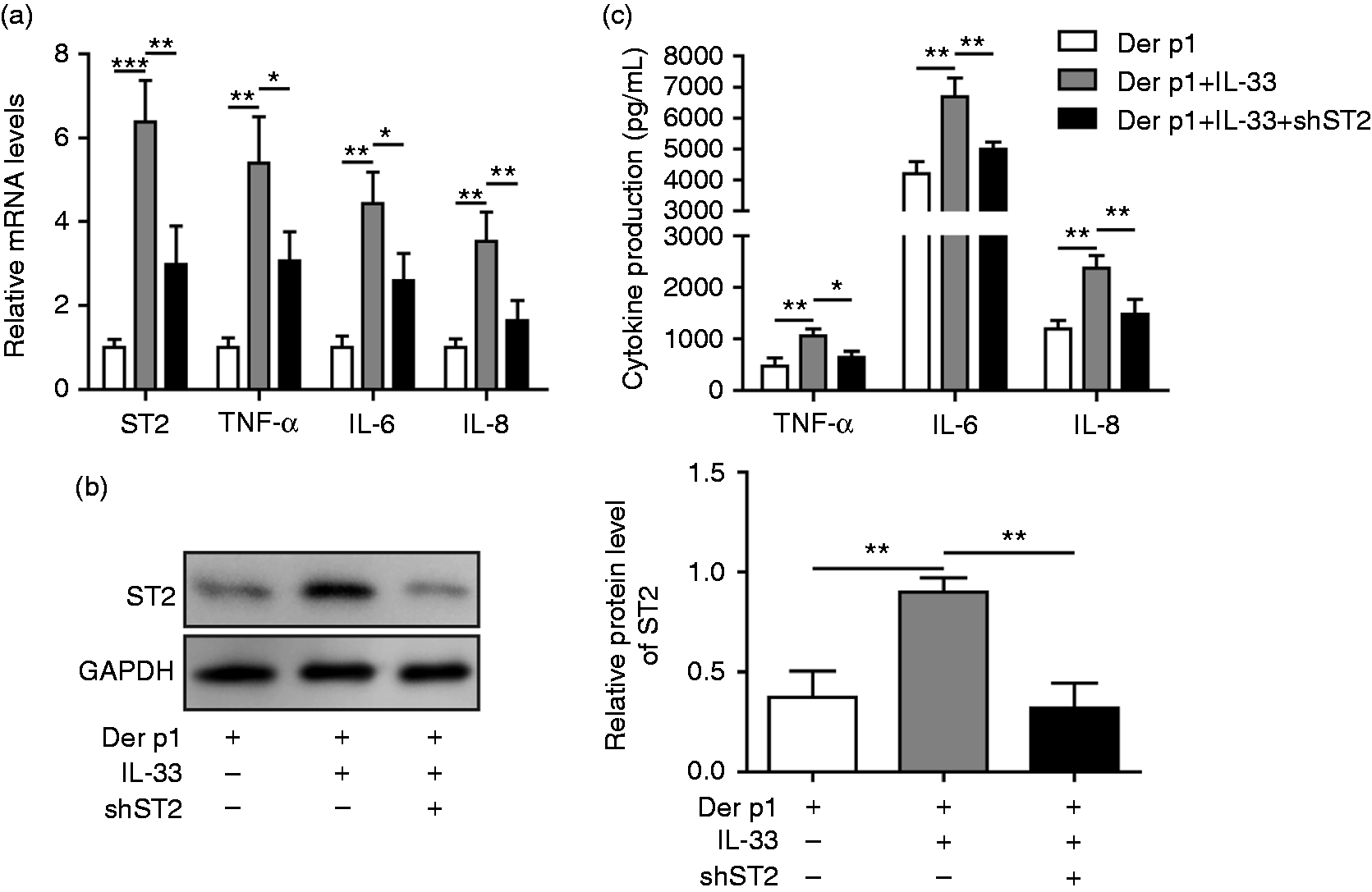
IL-33 mediates inflammation of nasal mucosal epithelial cells through ST2 receptor. (a) The mRNA levels of ST2, TNF-α, IL-6, and IL-8 were measured by qRT-PCR. (b) The protein level of ST2 was measured by Western blotting. (c) The concentrations of TNF-α, IL-6, and IL-8 were detected by ELISA. AR cells were divided into three groups: Der P1-treated, Der p1+IL-33 group, Der p1+IL-33+shST2 group. *P < 0.05 and **P < 0.01.
Sensitization of nasal mucosal epithelial cells promotes release of cellular inflammatory factors through the ERK1/2 pathway
To investigate the regulation mechanism of inflammatory factors in nasal mucosal epithelial cells, cells of the AR model were treated with the ERK1/2 pathway blocker, BVD-523. The data of qRT-PCR showed that the relative mRNA expression levels of IL-33 and ST2 were unchanged after treatment with an ERK1/2 blocker, while levels of inflammatory factors were decreased (Figure 4a) compared with cells of the AR model. As shown in Figure 4b, the protein levels of p-ERK1/2 and its downstream molecule p-RSK were increased markedly in cells of the AR model. However, the protein level of p-ERK1/2 stayed high and the p-RSK level was reduced in AR cells treated with BVD-523 (Figure 4b). The results of ELISA were consistent with the above results, indicating that protein expression levels of inflammatory factors were down-regulated after ERK1/2 blocker treatment (Figure 4c). These data suggested that nasal mucosal epithelial cells of the AR model promoted release of cellular inflammatory factors through the ERK1/2 pathway.
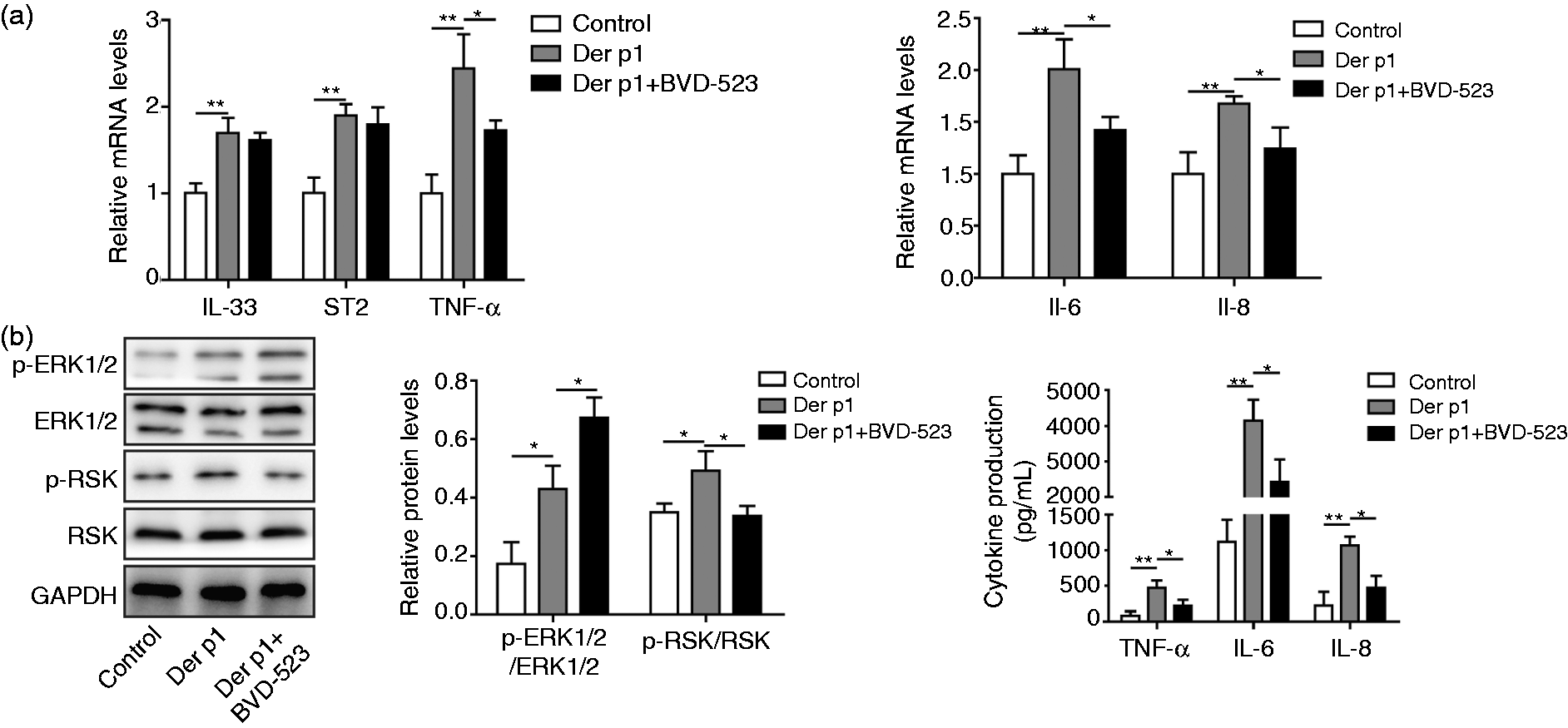
Sensitization of nasal mucosal epithelial cells promotes the release of cellular inflammatory factors through the ERK1/2 pathway. (a) The mRNA levels of IL-33, ST2, TNF-α, IL-6, and IL-8 were measured by qRT-PCR. (b) The protein levels of p-ERK1/2, ERK1/2, p-RSK, and RSK were measured by Western blotting. (c) TNF-α, IL-6, and IL-8 levels were detected by ELISA. Cells were divided into three groups: control group, Der P1-treated group, and Der p1+BVD-523 group. *P < 0.05 and **P < 0.01.
IL-33/ST2 promotes inflammatory response through ERK1/2 pathway
Finally, to further investigate the relationship between IL-33/ST2, ERK1/2 and cellular inflammatory factor release, ST2 was over-expressed by transfecting with pcDNA3.1-ST2. Cells of the AR model were treated with IL-33 or transfected with pcDNA3.1-ST2 in the presence of BVD-523. The data suggested that the expression levels of ST2 and inflammatory factors were further up-regulated in AR cells treated with IL-33 or pcDNA3.1-ST2 vector, but the expression levels of inflammatory factors were restored by BVD-525 treatment (Figure 5a). As assessed by Western blotting, p-ERK1/2 and p-RSK levels were significantly up-regulated in AR cells that were treated with IL-33 or transfected with pcDNA3.1-ST2, but the p-RSK level (but not the p-ERK1/2 level) was reversed in the BVD-525-treated group (Figure 5b). The trends of ELISA were similar with the results of qRT-PCR (Figure 5c). Collectively, the above results demonstrated that IL-33/ST2 promoted the inflammatory response through activating the ERK1/2 pathway.
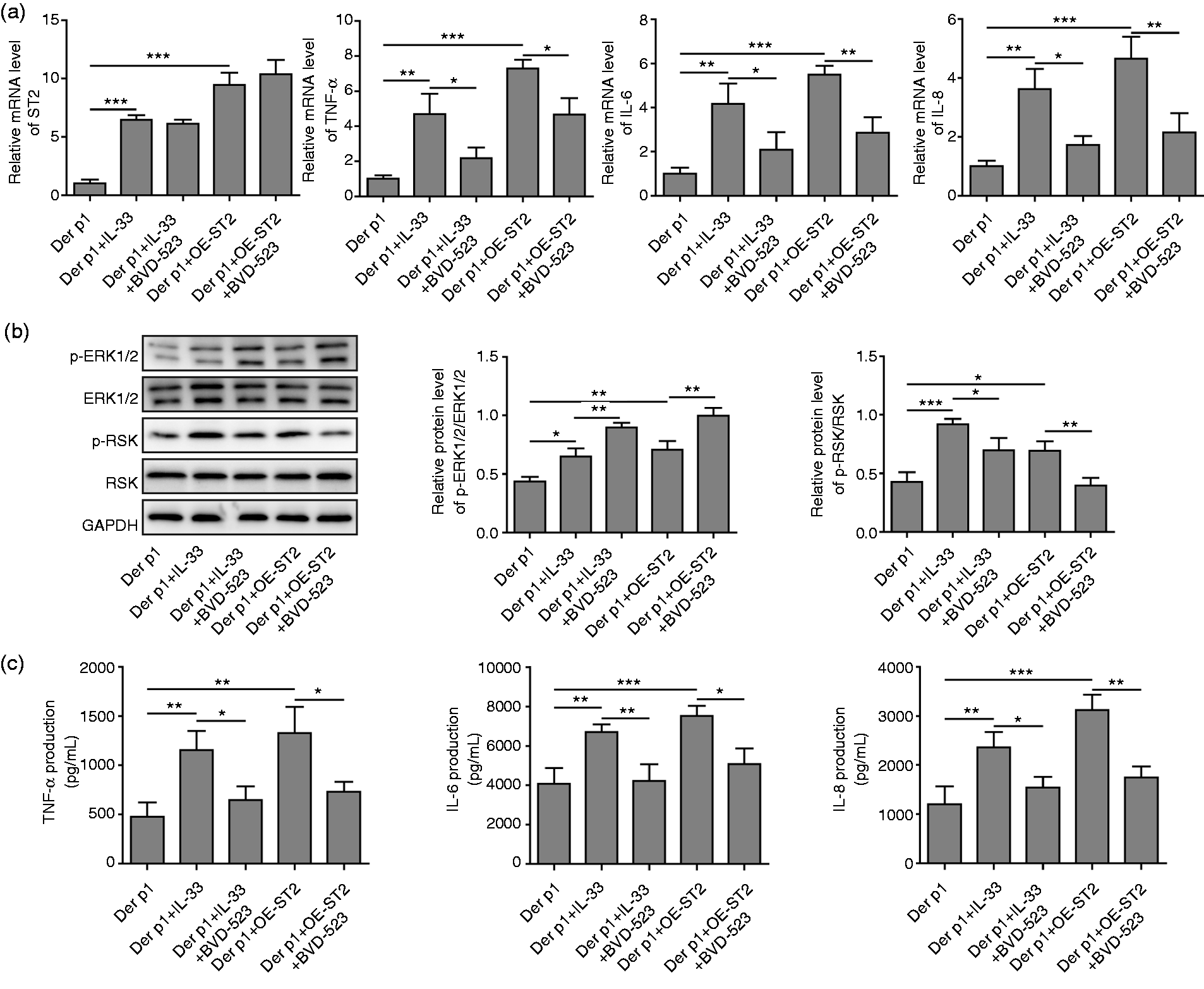
IL-33/ST2 promotes inflammatory response through ERK1/2 pathway. (a) The mRNA levels of ST2, TNF-α, IL-6, and IL-8 were measured by qRT-PCR. (b) The protein levels of p-ERK1/2, ERK1/2, p-RSK, and RSK were measured by Western blotting. (c) TNF-α, IL-6, and IL-8 concentrations were detected by ELISA. AR cells were divided into five groups: Der P1-treated group, Der p1+IL-33 group, Der p1+IL-33+BVD-523 group, Der p1+OE-ST2 group, Der p1+OE-ST2+BVD-523 group. *P < 0.05, **P < 0.01 and ***P < 0.001.
Discussion
AR is a common and frequently occurring disease in the clinic, and its pathogenesis has not yet been fully elucidated. 3 In recent years, with epithelial cells receiving increased attention in the pathogenesis of AR, epithelial cells have important immunoregulatory functions for both innate immunity and specific immunity. 18 Studies have shown that nasal mucosal epithelial cells are stimulated to produce corresponding cytokines that directly induce the body to produce type 2 natural lymphocytes and dendritic cells, promoting a Th2 type immune response and triggering local AR. 19 In the present study, we found that the IL-33/ST2 axis promoted inflammatory response of nasal mucosal epithelial cells via activating the ERK1/2 pathway in AR.
IL-33 is a recently discovered member of the new IL-1 family with dual functions of cytokine and nuclear factor. 20 IL-33 can be expressed continuously in various tissues and cells, especially in mucosal tissues in the respiratory tract, digestive tract, and other external tissues. 21 The release of IL-33 occurs mainly in injured or necrotic epithelial cells. 22 Sakashita et al. found that the level of IL-33 in serum of seasonal AR patients sensitive to Japanese cedar pollen was significantly higher than that of the normal control group, and that the increase of IL-33 was correlated positively with the degree of AR. 23 This finding suggests that IL-33 may play an important role in AR. Studies have found that exposure to allergens is essential for IL-33 release, after which the cytokines secreted by IL-33-stimulated Th2 cells induce goblet cell proliferation in the nasal mucosa. This hyperplasia may cause irreversible hypertrophy of the nasal mucosa, leading to the occurrence of AR. 24 As a cytokine, IL-33 binds to its specific receptor ST2 and induces the production of Th2 cytokines such as IL-6 and IL-8, thereby participating in Th2-related diseases such as AR and asthma. 6 Grotenboer et al. also pointed out that the genetic variation that associated with asthma at the IL-33 and IL-1 receptor-like 1 (IL1RL1) loci, is central to asthma pathogenesis. 25 Consistent with previous studies, our study showed that expression levels of IL-33, ST2, TNF-α, IL-6, IL-8, and IL-33, and cell apoptosis were increased after nasal mucosal epithelial cells were sensitized. Subsequently, our results further indicated that the expression of inflammation-associated cytokines were notably reversed after transfection with shIL-33 or shST2. IL-33 stimulation further resulted in the increase in both ST2 and inflammation-associated cytokines, and these effects were restored after shST2 treatment. Our data suggested that nasal epithelial cells mediated cellular inflammation via the IL-33/ST2 axis and IL-33 mediated inflammation of nasal mucosal epithelial cells through the ST2 receptor.
Similar to other IL-1 family members, the molecules downstream of IL-33 are mainly JAK2, JNK, MyD88, IRAK, RSK, p38 MAPK, and ERK1/2.26–28 Studies have found that the ERK pathway is not only involved in the regulation of cell responses induced by growth factors, but is also activated by TNF-α, IL-1, and others, and plays an important regulatory role in cellular responses induced by stimulation of stress and bacterial products.29,30 ERK1/2 is a key protein in the ERK pathway, and the ERK pathway-mediated intracellular signal transduction pathway is considered to be a classical MAPK signal transduction pathway. RSK, as a downstream molecule in the ERK pathway, can be activated by ERK to regulate intracellular signals, phosphorylate cytoplasmic and nuclear proteins participating in gene transcription, protein synthesis, cell differentiation and proliferation, and other processes. The ERK pathway involves a series of enzymatic reactions that ultimately modulate cellular activity and function through phosphorylation and dephosphorylation of ERK1/2. 12 ERK1/2 plays an important role in the inflammatory response and mediates several important inflammatory pathways.31–33 As a downstream effector molecule of IL-33, ERK1/2 is involved in the induction of inflammatory response in Th2 cells, mast cells, eosinophils, macrophages, and dendritic cells. 34 In this study, we showed that expression levels of inflammatory factors were down-regulated after ERK1/2 blocker treatment in the AR model. Furthermore, the expression levels of inflammatory factors induced by IL-33 stimulation or ST2 overexpression were reversed after ERK1/2 pathway blocker. In addition, p-ERK1/2 and p-RSK levels were significantly up-regulated in AR cells, but the p-RSK level was reversed after ERK1/2 pathway blocker. Our results indicated that nasal mucosal epithelial cells of the AR model promotes the release of cellular inflammatory factors through the ERK1/2 pathway and that the IL-33/ST2 axis affects cellular inflammatory factor release via the ERK1/2 pathway in nasal epithelial cells.
In summary, the IL-33/ST2 axis is a key signaling pathway in the development of AR, which activates the ERK1/2 signaling pathway in nasal epithelial cells. Therefore, interference with the IL-33/ST2/ERK1/2 signaling pathway may present new therapeutic options for AR.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a General Project of Science and Technology Plan of Science and Technology Department of Hunan Province (2013FJ6034) and the Project of Benevolence Fund of Hunan Provincial People's Hospital.