
Abstract
Sepsis, the 10th leading cause of death, is the most expensive condition in the United States. The immune response in sepsis transitions from hyperinflammatory to a hypoinflammatory and immunosuppressive phase; individual variations regarding timing and overlap between hyper- and hypoinflammation exist in a number of patients. While one third of the sepsis-related deaths occur during hyperinflammation, majority of the sepsis-mortality occurs during the hypoinflammatory phase. Currently, no phase-specific molecular-based therapies exist to treat sepsis. Coordinated epigenetic and metabolic perturbations orchestrate this shift from hyper- to hypoinflammation in innate immune cells during sepsis. These epigenetic and metabolic changes during sepsis progression and therapeutic opportunities they pose are described in this review.
Keywords
Introduction
Sepsis and septic shock are the leading causes of death in non-coronary intensive care units. Estimates indicate that nearly 5 million patients are diagnosed with this condition globally and over 200,000 patients die with sepsis in the United States alone each year; no specific therapies presently exist to treat these conditions.1,2 There is a dire need to find molecular based therapies to treat sepsis and septic shock. Sepsis transitions from early/hyperinflammatory to a late/hypoinflammatory phenotype; a number of co-morbidities determine the timing and persistence of any of these phases in septic patients. To further add to the variability of immune response, a small proportion of critically ill patients undergo a mixed hyper- and hypoinflammatory state, referred to also as persistent inflammation, immunosuppression, and catabolism syndrome (PICS). 3 PICS was first described in trauma patients. 4 Hyperinflammation during sepsis occurs concomitant with innate immune phagocyte cell (neutrophils and monocytes) activation of antimicrobial processes, which include oxidative metabolic sources of reactive oxygen and nitrogen species. The oxidative phenotype of sepsis is cytotoxic to innate and adaptive immune cells and specialized cells of vital organs as cell death pathways are activated. In response to that, the high energy consuming state of effector immunity/auto-toxicity rapidly transitions to a much lower energy demanding cytoprotective state in immune cells. This cytoprotective response, characterized by increased anti-inflammatory and decreased pro-inflammatory cytokine expression, is reflected in the phenomenon of endotoxin tolerance. 5 The nutrient substrates used to support pro-inflammation and pro-immune mechanisms are predominantly Glc and the Aa glutamine, which together support glycolysis and mitochondrial oxidative phosphorylation as ATP sources. 6 In contrast, lipids through lipolysis of both imported fatty acids and fatty acids stored as triglycerides support the energy conserving phenotype of mitochondrial β oxidation. Both, hyper- and hypoinflammatory sepsis-induced pathways profoundly depart from basal homeostasis, which must be restored if the death or prolonged sickness threats of the infection and the autotoxic byproducts of inflammation are to be avoided. Mechanistically, this means that anabolic antimicrobial control and catabolic tolerance control polarities must rebalance. Mechanistic insights into the transition of polarities from hyper- to hypoinflammation and pro-inflammatory to immune-repression are critical for understanding both cell and organism fate during sepsis and for designing molecular based treatments.
Mechanistically, the two categories of phenotypic shifts involved in reprogramming an immune response during the life threatening stress of sepsis are epigenetics and metabolism. Both epigenetics and metabolism depend on mitochondrial anabolic and catabolic bioenergy reprogramming. In this review, we describe how the epigenetic and metabolic pathways reprogram the innate immune response of cells during the sequential acute inflammatory response of life threatening sepsis and septic shock.
Hyperinflammation and hypoinflammatory programming of innate immunity in sepsis
Sepsis-3 defines sepsis as a “life-threatening organ dysfunction caused by a dys-regulated host response to infection” and septic shock as “subset of sepsis in which underlying circulatory and cellular/metabolic abnormalities are profound enough to substantially increase mortality.” 7 Ranked as the 10th leading cause of death sepsis is the most expensive condition in the United States with over $20 billion annual cost of care. 8 The host “resists” invading pathogen by mounting a systemic inflammatory response in innate immune cells such as macrophages and monocytes that produce pro-inflammatory cytokines, chemokines, and coagulation cascade within minutes. Further fuel to the fire of pro-inflammatory response is added by the activated pro-coagulant factors such as thrombin, Factor X, tissue factor via PAR1 signaling.9–11
However, hyperinflammation cannot be sustained, as it also attacks the host tissue and organs indiscriminately. The hyperactive immune cells transition to a deactivation/tolerance state also known as a “hypoinflammatory” and immunosuppressive phenotype. This phase is characterized by increased anti-inflammatory cytokine expression and decreased pro-inflammatory mediators biomarked by endotoxin tolerance (Figure 1).
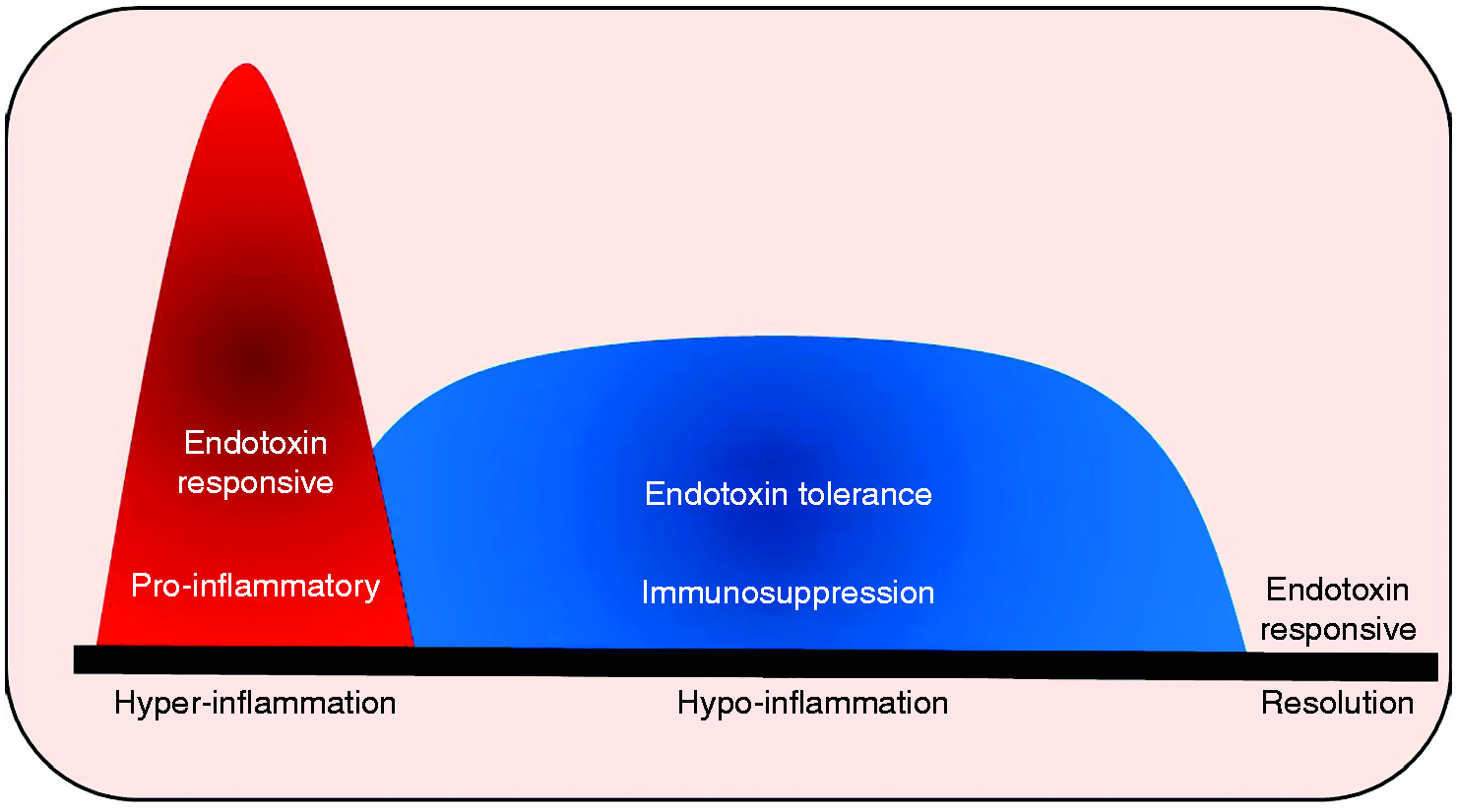
Immune response in sepsis: immune response in sepsis transitions from hyperinflammatory to a hypoinflammatory phase followed by resolution. Hyperinflammatory phase of immune cell activation is characterized by endotoxin responsive state with cytokine storm is cytotoxic to immune and other organ cells. Hypoinflammatory phase is characterized by endotoxin tolerance and immunosuppression. Hyper- and hypoinflammatory phases are associated with profound departure from homeostasis. Restoration of homeostasis is achieved during the resolution phase of sepsis.
In 1947 Beeson described endotoxin tolerance, 12 which is now defined as “reduced capacity of the host (in vivo) or of cultured macrophage/monocyte (in vitro) to respond to LPS activation following a primary intravenous stimulus.” 13 LPS binds to LPS-binding protein (LBP) and subsequently forms a complex with CD14 on cell plasma membrane; CD14 is anchored by phosphatidylinositol and does not have intracellular domain, 13 and a protein complex forms to signal danger. While receptors such as complement receptor type III, chemokine receptors, heat shock proteins have all been implicated in the past, since the discovery of TLR family, the research has mainly focused on TLRs, especially TLR4 as major receptor responsible for LPS signaling.13–16 Endotoxin tolerance is characterized by post receptor complex down-regulation of links to pro-inflammatory cytokine expression such as TNF-α, IL-1β, chemokine expression and up-regulation of anti-inflammatory IL-10 and TGF-β;17,18 LPS alters expression of thousands of genes in monocytes, which is gene class specific. 19 Thus, the mechanisms involved in endotoxin tolerance are complex and still expanding.20,21
The response to bacterial toxins seems to be highly dose-dependent. While “super low” doses of toxins elicit “priming,” “low” doses seem to induce endotoxin tolerance. 22 To that end, an important reaction to bacterial toxins known as the “Shwartzman reaction” is well described in the literature. Shwartzman first described this two-hit phenomenon in 1928 where the investigators administered toxic substance derived from Bacillus typhosus subcutaneously (first hit) in rabbits followed by intravenous injection of the same toxin (second hit). Approximately 2 h after the second hit, the subcutaneous-administration area showed inflammatory changes that progressively worsened and upon skin biopsy revealed hemorrhagic lesions. 23 While initially described as a local reaction, later it was discovered to be associated with systemic effects; prevented by glucocorticoid administration and human antiserum.24–26 The role of circulating leukocytes and platelets was noted later as well. 27 Interestingly, the higher concentrations of the first (preparatory) subcutaneous dose or the “first hit” of toxin was associated with lack of hemorrhagic lesions after intravenous toxin (second hit); however, the same concentration when used as a “second hit” was able to elicit the generalized Shwartzman reaction. 24 The investigators also noted “increased lactate concentration” in the subcutaneous tissue after the second hit, indicating increased glycolysis.24,26
Several studies in recent years have investigated Shwartzman and other “priming” phenomena. Evidence indicates that a “super low” dose of endotoxin (0.1 ng/ml range) leads to priming of innate immune cells via IL-1 receptor associated kinase 1 (IRAK1) via selective induction of
Cell models of monocytes/macrophages with in and/or ex vivo LPS stimulation in peripheral blood monocytes have provided mechanistic insights into endotoxin tolerance; however, this should always be confirmed in animal and human studies before assessing them as treatment targets. We delineated the hyper- and hypoinflammatory responses in vitro and then in vivo in a mouse model of sepsis.30,31 We used cecal ligation and puncture to induce sepsis, a model used since 1979. 32 We studied leukocyte adhesion in post-capillary venules as a “biomarker” for in vivo inflammation in the intestinal microcirculation. Leukocyte adhesion is a rate limiting step in inflammatory response, 33 but often overlooked in studies of introduction tolerance in cell models. We delineated three distinct phases. Within the first 12 h post-sepsis, a hyperinflammatory phase with leukocyte adhesion significantly increases in response to additional LPS stimulation in septic mice microvasculature, which is followed by a, hypoinflammatory phase in which leukocyte adhesion is tolerant to additional LPS stimulus. As a third phase, mice surviving for at least 72 h post-sepsis restore responsiveness to LPS as defined by adherence competence. 31 We observed that increased leukocyte adhesion assessed in vivo were associated with increased ICAM-1 and E-selectin adhesion molecule expression on the endothelial cells and P-selectin glycoprotein ligand, the ligand for the E- and P-selectin adhesion molecule expression on the circulating leukocytes. 31 These findings clearly support linear transition between sepsis hyper- and hypoinflammation in mouse sepsis, a paradigm also supported by cell and human sepsis models in monocytes. 30 It highlights the need to fully understand how the transition in phenotype programming is regulated.
Epigenetic reprogramming of innate immunity in sepsis
Epigenetics is the term first coined by Conrad Waddington in 1942, 34 which by current definition refers to a sustained environmental effects on gene expression program without change in the DNA sequence. 35 The epigenetic regulation of genes modifies the responsive euchromatin into reversibly silent heterochromatin that masks the transcription start sites by chromatin condensation. 36 Thus the epigenetic control, in general terms, revolves around winding and unwinding of the chromatin at specific gene set loci. Histones and their interactions with multiple transcription factors and cofactors package the DNA into variably accessible chromatin.37,38 As depicted in Figure 2, histone modifications on H2A, H2B, H3, and H4 tails control winding and unwinding of chromatin.
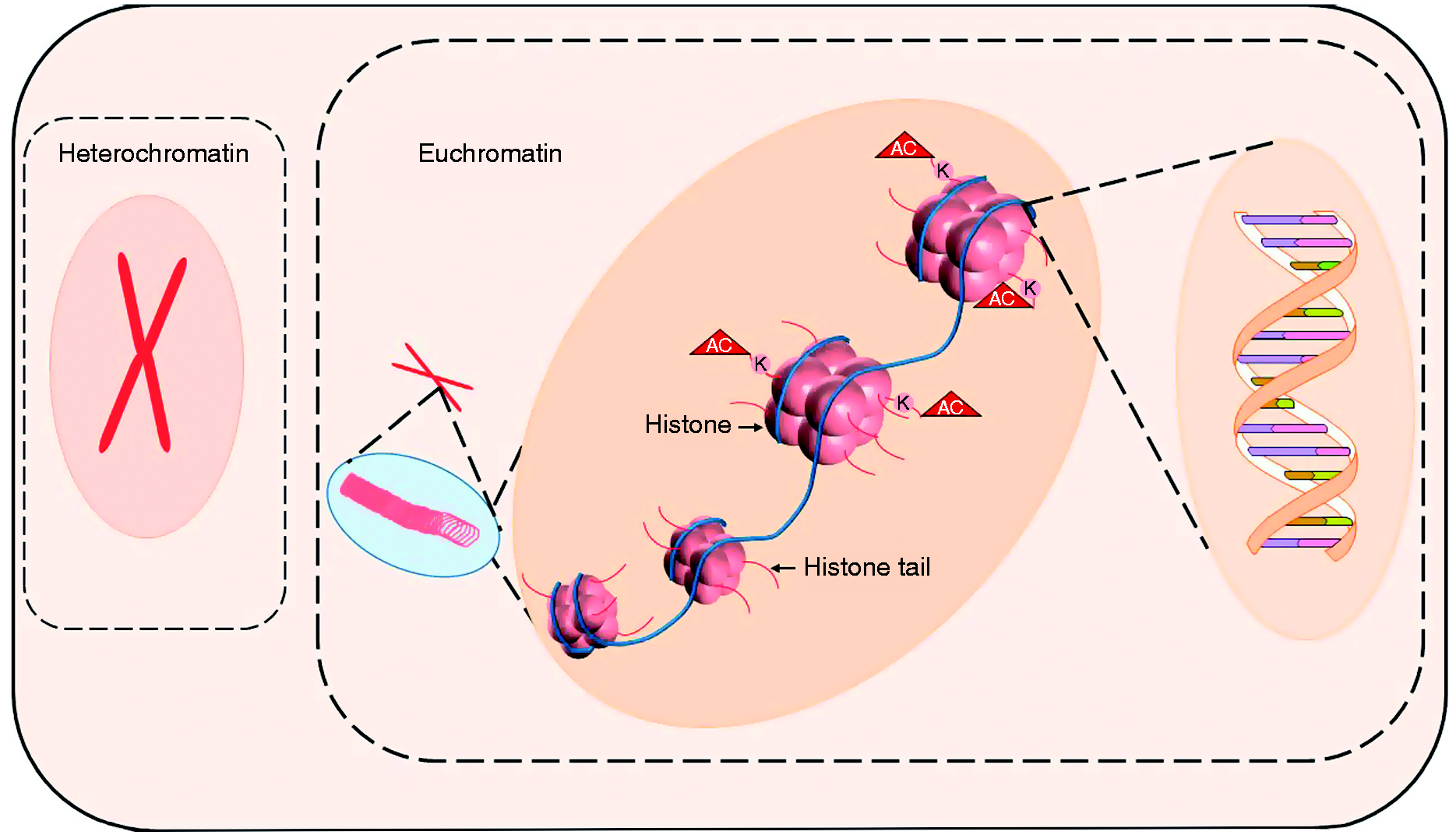
Epigenetic modifications: heterochromatin constitute of tightly packaged DNA around histone backbone, making DNA inaccessible to transcription factors. In response to cell signaling including stress, euchromatin formation (unwinding) occurs making DNA accessible for transcription factors. Several histone modifications on histone tails including acetylation, methylation, ubiquitination, and sumoylation modulate winding and unwinding of chromatin. Lysine (K) acetylation (AC) is mostly associated with euchromatin formation while methylation with silencing of DNA (not shown).
Histone modifications include acetylation, methylation, ubiquitination, phosphorylation, and sumoylation. 38 Histone acetylation predominantly supports gene transcription, and histone methylations play a dominant role in heterochromatin silencing of gene expression. How the histone tail modifications translate into euchromatin and heterochromatin formation is a complex but critically important network driving a sepsis outcome at the level of gene expression, as these epigenetic memory may provoke chronic disease. 38
Epigenetics of innate immunity hyperinflammation
During hyperinflammation, innate immune responses, intended to kill pathogens, enter a state characterized by excessive up-regulation of pro-inflammatory chemokines and cytokines that initiate and amplify systemic inflammation during sepsis. Included in the sepsis “cytokine storm” are these cytokines and chemokines arising from macrophages, dendritic cells and circulating neutrophils include cytokines such as TNF-α, IL-6, IL-1β, IL-12, IL-18, and IFN-γ, as well as chemokines such as CCL2, CCL3, CCL6, and CXCL8,39,40 expressed in blood and tissue monocytes macrophages and dendritic cells. 41 Pro-inflammatory cytokine expression is modulated by histone acetylation, supported by the observation that histone deacetylase inhibitor treatment suppress pro-inflammatory genes in response to LPS.38,41,42 Two antagonistic enzymes that control the acetylation status of chromatin are histone acetyltransferases (HATs) and histone deacetylases (HDACs), which transfer of acetyl molecule to and from acetyl-CoA to the lysines in the amino terminal region of histones. Evidence suggests that the highly positively charged N-terminal tails of histones can potentially interact with the negative phosphodiester backbone of the DNA. Major chromatin modifications associated with hyper- and hypoinflammation during sepsis are summarized in Table 1. Histone acetylation that functionally neutralizes a positive charge on specific lysines and thus loosening the chromatin structure to facilitate euchromatin formation. 43 Additionally, the newly acetylated lysines also act as the “molecular tags” for transcriptional activation. Thus, HATs are associated with euchromatin formation and transcriptional activation. 44 The availability acetyl-CoA is a rate limiting acetylation support of both transcription factors like NF-κB p65 and transacetylases like the P300 family that initiate epigenetic reprogramming of immune effector genes such as TNF-α, lL-1 b, and IFN-γ. An important concept associated with the early epigenetic reprogramming of acute inflammation in many cells including innate immune monocytes and macrophages is the “poised enhancer and promoter” concept, in which cell fate has been determined and cell function is rapidly responsive within euchromatin structure. 45 This allows TLR4 coupling to NF-κB and other pro-inflammatory signaling pathways to launch the acute inflammatory response, which in the case of sepsis is excessive or deregulated.
Chromatin modifications in sepsis induced inflammation.

Removal of repressor “methylation” marks on enhancer and promotor gene region also supports the pro-inflammatory phenotype in innate immune cells during activation. The histone 3 lysine 9 (H3K9me) repressive mark is erased during cell activation and restored during post-activation repression. 46 This demethylation requirement promoted DNA demethylase discovery. Jumonji domain-containing protein 3 (JMJD3) demethylase removes the trimethylation repressor mark on histone 3 lysine 27 (H3K27) during epigenetic chromatin modification. Fully differentiated macrophages functions are restricted from gene expression in response to environmental cues by H3K27 trimethylation of polycomb group (PcG) proteins, JmjC-domain H3K27me demethylase epigenetically derepresses the poised enhancer promoter state during acute inflammation. 47 Subsequently the effect of JMJD3 in derepression was shown to be independent of its demethylase activity. 48 Other demethylases, such as lysine-specific histone demethylase 1A (LSD1) also known as lysine (K)-specific demethylase 1A (KDM1A) are derepressors of acute inflammation and modulation of LSD1 shows therapeutic promise in endotoxic shock. 49 Better understanding of the epigenetic pathways responsible for derepressing inflammation and immunity is an important opportunity for drug discovery.
Epigenetic reprogramming of innate immunity hypoinflammation
The hypoinflammatory phenotype of sepsis and its endotoxin tolerance biomarker epigenetically transition high energy consuming state of early sepsis to a much lower energy state, which in some patients in animal models simulates the status of hibernation, suspended animation and energy. 50 A likely and ill-fated consequence of the low energy and hypoinflammatory state with endotoxin tolerance is sepsis-induced profound immunosuppression. This potentially lethal phenotype dominates sepsis clinically and is responsible for more deaths that microvascular collapse during hypoinflammatory sepsis. 51 The importance of this undesirable sepsis associated phenotype is evidenced by sustained an opportunistic infection in humans and animals with sepsis. 52
Others and we have reported epigenetic regulation involved in endotoxin tolerance of innate immune monocytes and macrophages during hypoinflammation is associated with a switch in chromatin status from euchromatin to facultative heterochromatin (Figure 2) formation.36,53 This switch requires repositioning of nucleosome and concomitant formation of protein complex responsible for transcription repression, which, in turn, requires a switch from NF-κB p65 transcription factor to RelB.54–57 A clinically relevant and often overlooked observation is that the process of formation of euchromatin and switch to heterochromatin occurs within hours of inflammatory stimulus to an innate immune cell and activation of TLR4.
During the silencing and endotoxin tolerance, NF-κB factor RelA/p65 trans activator is replaced by NF-κB factor RelB. RelB represses a set of genes and supports formation of heterochromatin while activating euchromatin on other sets of genes thus supporting activation. 54 The heterochromatin formation is associated with alteration of histones in the proximal promotors of pro-inflammatory genes. This occurs via formation of protein complex consisting of G9a transmethylase that dimethylates H3K9, creates a platform for HP1 binding, leads to the recruitment of the DNA methyl transferase Dnmt3a/b and increases promoter CpG methylation, thus forming a stable epigenetic repressor complex.54,58 Decreased acetylation on histone 4 (AcH4) in addition to trimethylation of lysine 4 on histone 3 (H3K3me3) is supported in endotoxin tolerant pulmonary macrophages in sepsis mice by up-regulation of IRAK-M. 53
We reported that NAD+-dependent class III HDAC family of proteins, sirtuins are crucial to the transition from hyper- to hypoinflammatory response in sepsis.30,31,59 The critical difference in the SIRTs and other HDAC is their dependence on NAD+ and their regulation of both immunity and metabolism. There are seven mammalian homologs of sirtuin proteins (SIRT1-7). We have shown that SIRT1 and SIRT2 directly deacetylate NF-κB p65 to deactivate it during hypoinflammatory phase of sepsis; SIRT1 and SIRT2 inhibition during this hypoinflammatory phase not only reverse endotoxin tolerance, but also improve mortality in septic mice.31,60,61 In addition to modulating the NF-κB p65 acetylation, sirtuins also direct and work together with the repressor complex mentioned above. In a two-way relationship, sepsis modulates redox-sensitive cysteines; increased sirtuin oxidation during hyperinflammation and decreased oxidation during hypoinflammation.62,63 Thus, the sirtuin family of proteins are epigenetic, posttranscriptional and posttranslational guardians of homeostasis. They sense and regulate the coordination of redox and intermediary metabolism substrate selection for energy control. In addition, they direct the innate and adaptive immune response. Combined and coordinated molecular controls over all of these networks in mice and their homeostasis rheostat property are supported by the results of our treatment studies.31,60,64 This raises the question whether flexible metabolic fuel selection directs innate immunity during sepsis.
Metabolic reprogramming of innate immunity in sepsis
Growing evidence supports that the metabolic shifts are crucial for temporally changing the hyper- and hypoinflammatory innate immune cell phenotypes in inflammatory phenotype during sepsis. Studies in cell models of sepsis and isolated leukocytes from sepsis patient samples suggest that these shifts occur during sepsis-inflammation. Furthermore, the metabolic shifts may precede the immune cell phenotypic phase shifts.31,65
Metabolic reprogramming of hyperinflammation
During hyperinflammatory phase of sepsis, the innate effector cells are tasked with pathogen clearance, a process that consumes large quantities of energy in the form of ATP. This energy is needed for differentiation as well as immune effector microbial accounting processes. The innate immune cells have three specific requirements: (1) high energy, (2) activating effector immunity, and (3) rapid cell regeneration. A major source of ATP in monocytes/macrophages/dendritic cells during extreme stress is glycolysis. In innate immune cells during hyperinflammation, a switch to aerobic glycolysis or “Warburg effect” common to cancer cells achieves this, as depicted in Figure 3a. Otto Warburg described this phenomenon when cancer cells preferentially utilize glycolysis to provide substrate for nucleotide synthesis for regeneration and ATP requirement to sustain proliferation. 66
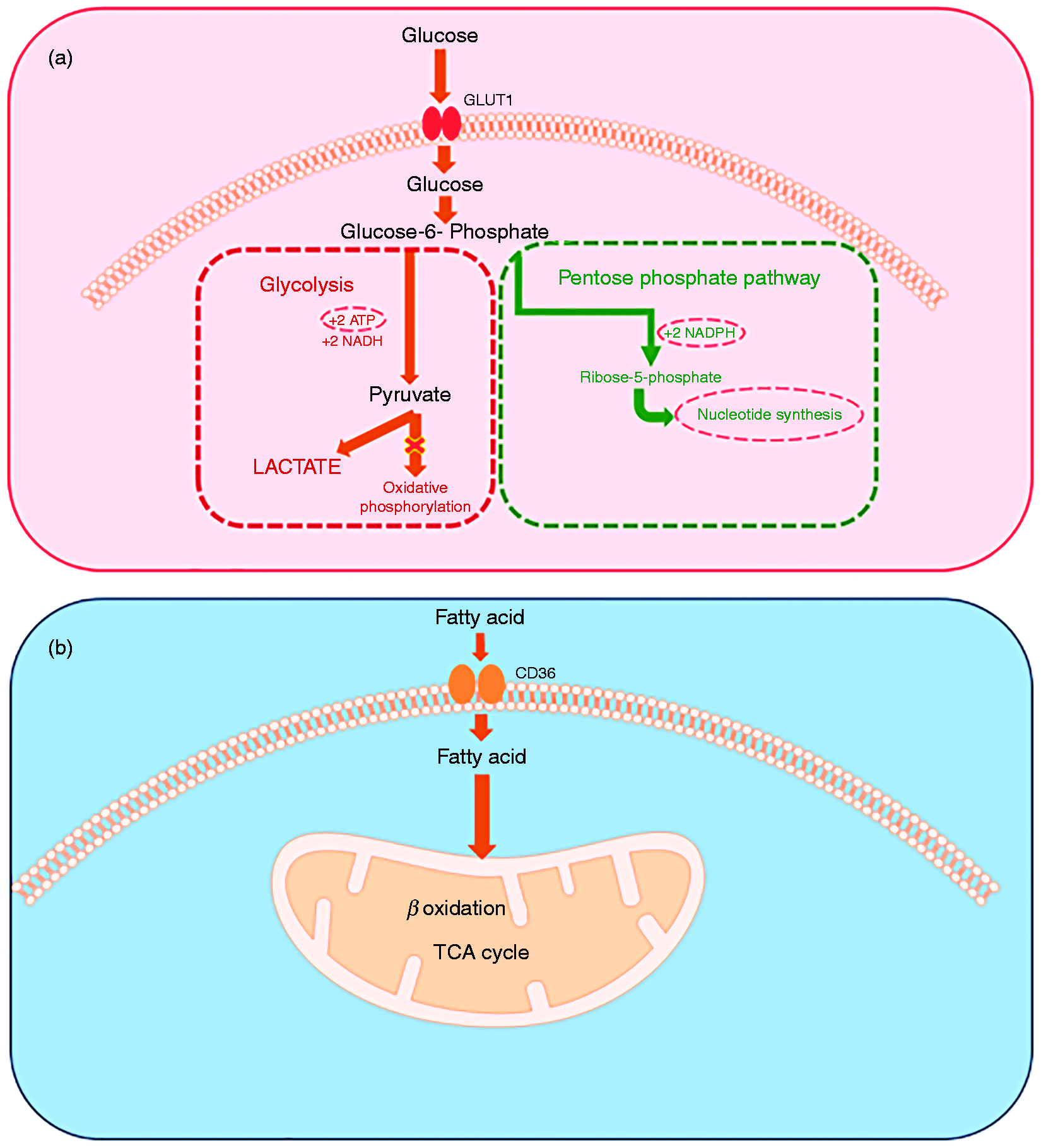
Metabolic changes of during sepsis. (a) Aerobic glycolysis by immune cells during hyperinflammatory phase of sepsis. During this phase, there is inhibition of oxidative phosphorylation and selective increase in PPP. Aerobic glycolysis provides ATP generation fulfilling the “high energy” demand of cells. In addition, there is generation of NADPH and Rib-5-phosphate to fulfil the “activation of effector immunity” and “cell regeneration” to support pathogenic killing, as detailed in the text. (b) The fatty acid uptake during the hypoinflammatory and cytoprotective response of sepsis. This is associated with increased CD36 expression on immune cell surface. Hypoinflammatory phase is associated with increased β-oxidation and tricarboxylic acid (TCA) cycle to produce energy for cell survival.
High energy
Innate immune cells, the first responders of the inflammatory stimulus mobilize to phagocytose and kill the invading pathogen. They are mobilized in response to the inflammatory stimulus by consuming energy available to cell from Glc carbon combustion.67–69 Disrupting glycolysis has profound adverse effects on phagocytosis, but disrupting mitochondrial-Glc oxidation does not.68,70 Glc enters the immune cells via up-regulated GLUT1 expression in response to an inflammatory stimulus.71–73 Once inside the cell, Glc converts to pyruvate during multistep glycolysis. 65 Pyruvate is either converted to lactate and is secreted rapidly, or enters mitochondria after decarboxylation by pyruvate dehydrogenase complex (PDC) to acetyl-CoA. Intramitochondrial acetyl-CoA can enter the tricarboxylic acid cycle and electron transport chain in the mitochondria, while cytosolic acetyl-CoA fuels fatty acid synthesis. The extramitochondrial glycolysis nets only two molecules of ATP, while the total yield of ATP molecules from the extra- and intramitochondrial Glc oxidation is 36 molecules of ATP. However, while highly efficient in regard to ATP generation, oxidative phosphorylation is a slow process while glycolysis that can be up-regulated extremely rapidly and meet the demand for ATP generation quickly. Thus, the pathogen-fighting innate immune cells use glycolysis preferentially over the oxidative phosphorylation to meet the high energy life threatening microbial invasions by increasing Glc flux and glycolysis to lactate. In fact, the oxidative phosphorylation is inhibited in immune cells during acute stress from infection.66,74–77
Activating effector immunity
Innate immune cells use “weapons” to kill or contain invading microbes. Reactive oxygen species (ROS) promote pathogen killing.78–80 Aerobic glycolysis seen in the innate immune cells during hyperinflammation along with the inhibition of oxidative phosphorylation leads to accumulation of Glc-6-phosphate; Glc-6- phosphate then feeds into the pentose phosphate pathway (PPP) which in addition to the formation of Rib-5-phosphate, supports generation of NADPH molecules and in turn generation of NADPH oxidase, a ROS, or the “killing capacity” much needed by these cells.65,81,82 Glc-6-phosphate dehydrogenase (G-6PD), a key regulatory enzyme of PPP, is essential for neutrophil extracellular trap (NET) formation to further assist with phagocytosis of pathogens. Furthermore, G-6PD deficiency is associated with increased susceptibility and mortality in sepsis, likely due to decreased phagocytosis with impairment of PPP and NADPH activity.81,83–86
Rapid cell regeneration
Apoptosis with profound lymphopenia occurs early and persists during sepsis with 10–20% of cells dying;87,88 while exact mechanism/mechanisms remain unclear, many overlapping cells death mechanisms likely occur concomitantly. Accordingly, there is a need for continued regeneration of immune cells to continue effective phagocytosis, pathogen killing and inflammation resolution. Rapid regeneration and differentiation of immune cells requires broad increases in biomass. The PPP generates Rib-5-phosphate, a substrate for nucleotide synthesis, to support the much-needed cell generation during early sepsis. Thus, once again, the rapid regeneration of immune cells, much like that of cancer cells utilizes “Warburg effect” to fulfil the biomass requirement. 65 Glycolysis also fuels a lipid synthesis and promotes protein synthesis from Aas, and RNA and DNA synthesis by one carbon glycolysis support of serine glycine and synthesis. Thus, during the hyperinflammatory phase of sepsis, aerobic glycolysis at least partially fulfils all the three requirements of the immune cells to resist uncontrolled systemic infection.
The hyperinflammatory response is a double-edged sword; while resisting infecting organism, the cells and organs must protect themselves from cytotoxicity arising from the “effector response.” To do this, innate and adaptive immune cells and selective organ cells such as kidney epithelium, endothelium, intestine villi, and hepatocytes invoke evolution’s two survival principles of resistance and tolerance. At the metabolic level, the infection-resistance and auto-toxicity are in conflict during sepsis: immune resistance during hyperinflammation requires anabolism and its support of increased biomass, but tolerance during hypoinflammation requires a catabolic low energy source provided by oxidizing fatty acids, and may even need to enter a state of anergy or suspended animation.
Metabolic reprogramming of hypoinflammation
Profound depletion of energy during hyperinflammation cause immune cells to enter “cell hibernation” and abandon resistance to infection during hypoinflammatory response of late sepsis. 50 This transition from hyper- to hypoinflammatory response occurs in both, the innate and adaptive immune cells.89,90 The hypoinflammatory phase resembles suspended animation or hibernation characterized by mitochondrial dysfunction and resultant perturbations in cellular metabolism. Evidence supports that a hypometabolic state replaces a more transient hypermetabolic anabolic state of cell, animal, and human models of sepsis. Sirtuins play a crucial role in this switch from hyper- to hypometabolic state concomitant with the switch from hyper- to hypoinflammatory response. Mitochondrial SIRT3, which is controlled by SIRT1 promotes increased β-oxidation and Aa anaplerosis as mitochondria-driven catabolism. 91 This major shift is controlled by both AMPK disruption of mTOR-dependent protein synthesis and combined sirtuins 1, 3, and 6 response in support of the catabolic energetics, which together drive endotoxin tolerance and promote innate immune suppression.71,91,92 Recent data also suggest that SIRT1 may act through dendritic cells to increase immune repressor cell function and decrease CD4+ T cell pro-immune responses. 93 If confirmed, this supports the notion that innate and adaptive immune competence are coordinated during sepsis by changes in metabolic substrates that reciprocally fuel anabolism and catabolism. Interestingly, the anabolic, excessively oxidative state of hyperinflammation is countered by hypoinflammatory response with the support of antioxidants such as glutathione, superoxide dismutase, and thioredoxin. High levels of ROS directly inactivate cysteines in the zinc tetrathiolate motif of sirtuins; mechanistically, this is highly relevant for fine-tuning of inflammatory response of sepsis. Specifically, a direct and reversible cysteine thiol oxidation on SIRTs 1, 2, and 6 derepresses pro-inflammatory genes enabling hyperinflammation and anabolism.62,63,94
Nutrients and oxygen consumption decrease in muscle and innate immune cells during sepsis in animals and humans.95,96 If the biomass expansion and cell regeneration were to continue in the face of decreased oxygen and ATP supply, the resulting energy deprivation would massively activate cell apoptosis. 97 Perhaps the hibernation and suspended animation phenotypes of sepsis hypoinflammation are survival necessities that counter death pathways by a “switch off” from high energy to low energy. 95 Support for this concept and for metabolic mechanisms underlying sepsis outcomes during tolerance are mounting.
We and others found that innate immune monocytes and macrophages, obtained from the hypoinflammatory phase from different models of sepsis, change their energy source substrate selection from Glc-fueled glycolysis to fatty acid oxidation,71,91 as depicted in Figure 3b. Netea et al. reported broad defects in both glycolysis, fatty acids metabolism, and mitochondrial bioenergetics in human and rodent blood monocytes with highly lethal septic shock. 39 The investigators also found that this energy deficit-state with immunoparalysis of blood monocytes could be partially reversed by interferon γ treatment of human sepsis patients or their monocytes. 92
Our studies of lipid metabolism during the hypoinflammatory phase differed in that CD36 plasma membrane fatty acid importer and CPT-1 mitochondrial membrane importer of acyl carnitine derived fatty acids increased. 71 This difference may be related to dynamic shifts in nutrient sources and metabolism in monocytes and macrophages during sepsis. Reports also support that macrophages undergo alternative M2 phenotype activation under support of STAT6 and PGC-1, thus directly linking oxidative phosphorylation with anti-inflammatory response; 98 notably, sirtuin 1, lies proximal to PGC-1 during hypoinflammatory phase of sepsis. 71
The role of metabolism in directing the course of sepsis is growing as well. We found that the catabolic phenotypes are controlled by increased expression pyruvate dehydrogenase kinases, which maintains an inflexible tolerance-phenotype by precluding a reversal of catabolic energetics to anabolic energetics balance. As a result, cell regeneration processes might arrest. 99
Although indirect support of the innate immunity immunometabolism concept, a large non-biased metabolomics study of plasma from sepsis patients and non-human primates suggests a relationship between fatty acid oxidation pathway and sepsis-survival.100,101 The studies indicate that dys-regulation of fatty acid oxidation pathway and accumulation of short and long chain acyl carnitine fatty acids early in the course of the disease predict sepsis-survival. Specifically, survivors showed increased levels of six carnitine metabolites while 16 carnitine esters increased in non-survivors of sepsis. In this study as well, fatty acid CPT1 transporter levels decreased in sepsis non-survivors.100,101
Epigenetic and metabolic targeting during sepsis
Epigenetic and metabolic reprogramming in sepsis pose several opportunities for therapeutic targeting. Several such targets have been investigated over the past decade.
Epigenetic targets
Histone deacetylase inhibition used prior to sepsis induction in mice with sepsis is associated with increased survival via attenuation of hyperinflammatory response.102,103 We have shown that sirtuin overexpression during the hyperinflammatory phase of sepsis also increases survival in rodent sepsis. Others and we have shown that resveratrol pre-treatment in sepsis is associated with increased survival in mice with sepsis via attenuation of hyperinflammatory response.61,104–106 Interestingly, an old drug procainamide is also shown to be effective in attenuating hyperinflammation and short-term survival in endotoxemic rats via inhibition of DNA methylation. 107
Increased sirtuin expression not only is critical to switch from hyper- to hypoinflammation, but also for sustained hypoinflammatory response in rodent sepsis. Accordingly, we showed that sirtuin inhibition during the hypoinflammatory phase of sepsis not only reverses hypoinflammatory response but also improves survival in rodent sepsis. Sirtuin regulation during hypoinflammatory response of sepsis seems to depend on biological context. To that end, we showed that SIRT1 inhibition in lean and SIRT2 inhibition in obese mice during the hypoinflammatory phase are associated with significant increase in survival.31,59
Metabolic targeting
As discussed earlier, there is decreased Glc utilization by immune cells during the hypoinflammatory phase of sepsis. We have shown recently that PDC plays a critical role in modulating this response; moreover, inhibition of pyruvate dehydrogenase kinase using dichloroacetate (DCA) reactivates PDC, increases mitochondrial oxidative bioenergetics in immune cells and hepatocytes, promotes immune, and organ homeostasis. In addition, we also showed that DCA accelerates bacterial clearance and improved survival in mice with sepsis. 99 DCA is already in clinical trials for other disease processes, the clinical use of DCA in sepsis patients remains to be studied.
Challenges to therapeutic targeting
While various exciting metabolic and epigenetic targets are studied in pre-clinical models of sepsis, several roadblocks continue to exist before the true translational potential of these targets can be studied in sepsis patients. One of the main roadblocks is recognition of the exact phase the patient belongs to at any given time. Sepsis is a dynamic disease; the immune response in sepsis patients transitions from hyper- to hypoinflammation. This transition of phase remains elusive in sepsis patients. There is a dire need for a single biomarker or a panel of biomarkers that can be rapidly tested and available for use in patients before employing these phase-specific targets; one example being sirtuin up-regulation during hyperinflammation while sirtuin blockade during the hypoinflammatory phase of sepsis. These biomarkers need to take the epigenetic and metabolic changes in the innate immune response into account.
Conclusions
The immune response in sepsis transitions from a hyperinflammatory to a hypoinflammatory phenotype. Epigenetic and metabolic changes cooperatively drive this polarity in immune and non-immune organ cells and tissue, providing opportunities for therapeutic targeting, as summarized in Figure 4. Developing biomarkers to identify the hyper- and hypoinflammatory kinetics is urgently needed to guide opportunities for molecular targeting in sepsis.
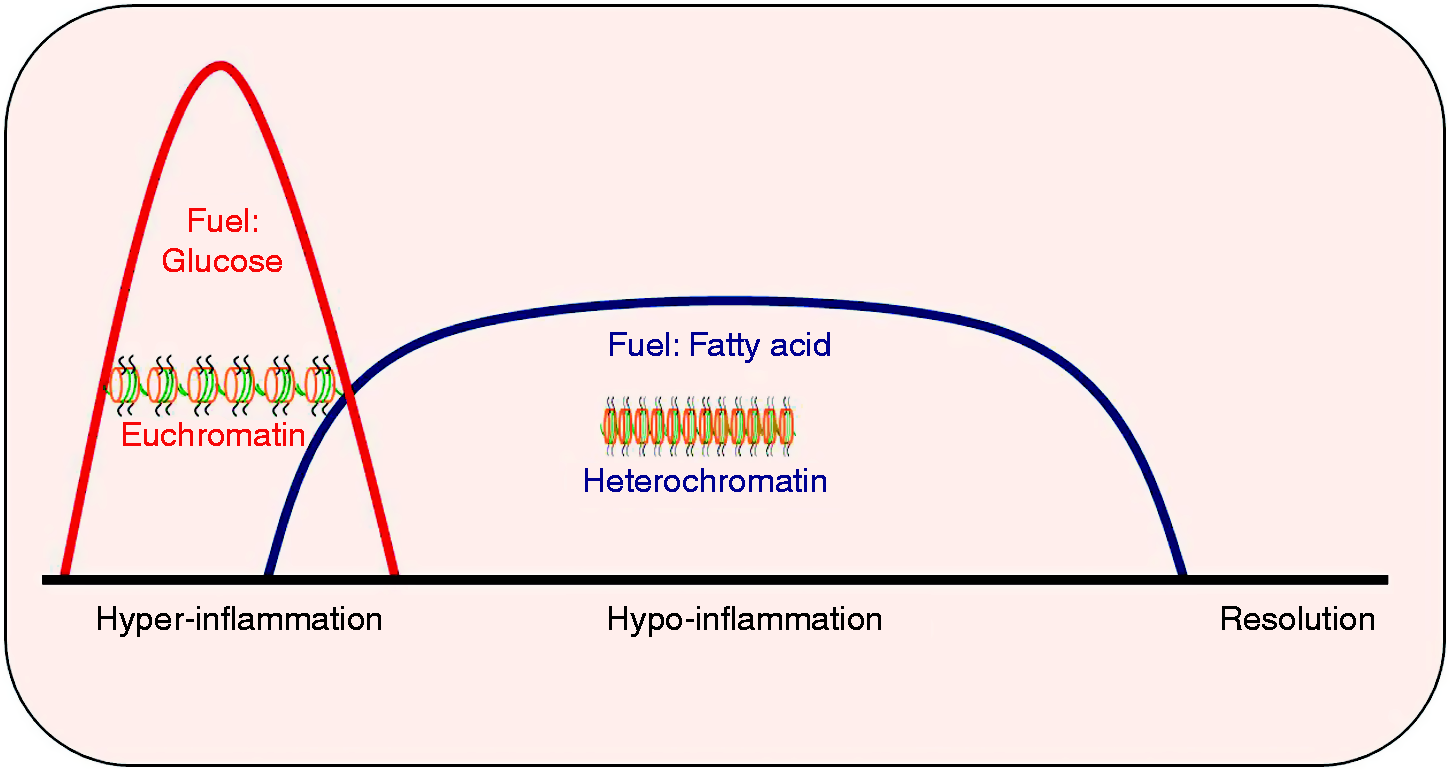
Summary of epigenetic and metabolic changes of sepsis: epigenetic and metabolic changes coordinate to change hyper- to hypoinflammatory phase in immune cells.
Footnotes
Authos’s note
Vidula Vachharajani is now affiliated with Critical Care Medicine/Respiratory Institute, Inflammation and Immunity/Lerner Research Institute, Cleveland, OH.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health (Grant Numbers R01GM099807, R01AI065791, R01AI079144, and 1R35GM126922).