
Abstract
This study investigates the modulation of Type I IFN induction of an antiviral state by HIV. IFNs, including IFN-α, are key innate immune cytokines that activate the JAK/STAT pathway leading to the expression of IFN-stimulated genes. IFN-stimulated gene expression establishes the antiviral state, limiting viral infection in IFN-α-stimulated microenvironments. Our previous studies have shown that HIV proteins disrupt the induction of IFN-α by degradation of IFN-β promoter stimulator-1, an adaptor protein for the up-regulation and release of IFN-α into the local microenvironment via the retinoic acid-inducible gene 1-like receptor signaling pathway. However, IFN-α is still released from other sources such as plasmacytoid dendritic cells via TLR-dependent recognition of HIV. Here we report that the activation of the JAK/STAT pathway by IFN-α stimulation is disrupted by HIV proteins Vpu and Nef, which both reduce IFN-α induction of STAT1 phosphorylation. Thus, HIV would still be able to avoid antiviral protection induced by IFN-α in the local microenvironment. These findings show that HIV blocks multiple signaling points that would lead to the up-regulation of IFN-stimulated genes, allowing more effective replication in IFN-α-rich environments.
Introduction
Despite four decades of HIV research, there is no a functional cure for HIV. HIV is the cause of AIDS, which remains one of the world’s major pandemics. By 2015, there were around 37 million people living with HIV across the globe. The current effective treatment for HIV is highly active antiretroviral therapy, which slows down the progression to AIDS by inhibiting HIV replication. However, eliminating all the virus from the host still proves difficult as HIV often persists latently in the host genome and will reactivate, restarting the pathway towards AIDS without continuous antiretroviral therapy.
HIV mainly infects CD4+ T cells that express CD4+ receptors and CCR5 or CXCR4 co-receptors on the cell surface. Along with virus-induced cytopathicity, infected CD4+ T cells are also gradually eliminated and depleted by host immunity to prevent further infection and protect healthy cells. Protective mechanisms occurring at the level of innate immunity include antiviral responses involving viral recognition, release of cytokines, activation of macrophages and natural killer cells, etc. 1 PRRs such as TLRs and retinoic acid-inducible gene 1 (RIG-I)-like receptors (RLRs) recognize HIV-infected cells and signal downstream to turn on the antiviral state against HIV. HIV nucleic acids produced during infection of target cells are recognized by RLRs in the cytoplasm of the infected cells. 2 Additionally, macrophages and plasmacytoid dendritic cells (pDCs) recognize HIV-infected cells via TLRs, particularly TLR7 and 9.3,4 Both these recognition events result in signaling that eventually leads to the induction of Type I IFN, such as IFN-α and IFN-β. While IFN-β can be released by a majority of non-immune cells, IFN-α , which consists of 13 subtypes, is a cytokine that is often released by immune cells and signals and guides innate immunity.5,6 For example, pDCs can released IFN-α 1000-fold higher than any other cell type in the immune system. 7
IFN is well known to activate the antiviral state of innate immunity by up-regulating the expression of IFN-stimulated genes (ISGs) via the JAK/STAT pathway. 8 This pathway is activated following the binding of IFN-α by the heterodimeric IFN-α/β receptor (IFNAR), which orchestrates the phosphorylation of STAT1 and STAT2 via the Tyk2 and Jak1 kinases. 9 Phosphorylated STAT1 (pSTAT1) and pSTAT2 form a complex with IFN regulatory factor-9 (IRF9) to become a transcriptional activator that is designated IFN-stimulated gene factor 3 (ISGF3). ISGF3 enters the nucleus and binds to IFN-stimulated response element (ISRE) within the promoter region of ISGs. The bound ISGF3 activates the transcription of hundreds of ISGs. Multiple ISGs, including apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3G (APOBEC3G) and tetherin, are important in controlling HIV replication in infected cells. APOBEC3G interferes with viral replication and tetherin inhibits the release of virions from infected cells.10,11 Recently, others have suggested that STAT3 may also be key in these pathways. 12
Patients infected with HIV progress to AIDS around 10 yr after the initial infection by HIV if not treated, even with the many facets of anti-viral immunity present in a host. One reason for such persistent replication in the face of immune activation is that HIV bypasses multiple antiviral responses, with HIV proteins able to disrupt the functions of various steps in establishing the IFN-induced antiviral state. Upstream of IFN production, RLR signaling is interrupted by Vpu and Nef as they destabilize the RLR adaptor IFN-β promoter stimulator-1 (IPS-1) and protease specifically cleaves RIG-I, further ablating the induction of IFN by viral RNA recognition.13,14 In addition, some reports suggest that Vpr and Vif block IRF3 phosphorylation, compounding the inability of an HIV-infected cell to produce IFN upon infection. 15 Consequently, infected cells are less efficient in controlling HIV replication due to their inability to produce IFN-α, leading to less signaling for up-regulating ISG production. Additionally, a handful of IFN-induced ISGs have been reported to be targeted by HIV to inhibit their particular function. For example, Vif is known to down-regulate APOBEC3G and Vpu is proposed to reduce the expression of tetherin.16,17 Although these mechanisms inhibit IFN production in HIV-infected cells and disrupt a small handful of ISG from the more than 300-member pantheon of ISGs, there seem to be additional mechanisms in place to disrupt HIV.18,19
However, patients that are acutely infected with HIV and progressing to full-blown AIDS have been clinically documented to have a high level of serum cytokines present, including IFN-α.20,21 Serum IFN-α is primarily induced by pDC recognition of infected cells via TLR-dependent pathways. Given excessive IFN-α release by pDCs into the serum and local microenvironments within an individual infected with HIV and the effectiveness of many ISGs to contain the virus, a conundrum exists to understand why the levels of ISGs in the CD4+ T cells of a person infected with HIV are not sufficient to control HIV replication. 22
HIV must have other mechanisms to down-regulate the expression of ISGs to replicate in an IFN-rich environment beyond a targeted approach to certain ISGs. Therefore, we hypothesize that HIV proteins block ISG expression at the JAK/STAT signaling pathway even in the presence of exogenous IFN-α. In this way, HIV can stop the entire collection of ISGs that may induce direct anti-viral mechanisms as well as indirect changes in cellular metabolism that make it difficult for the virus to replicate efficiently. Here we show that HIV can directly block IFN-α-induced JAK/STAT signaling by inhibiting the phosphorylation of STAT1. This inhibition will lead to the reduced expression of all ISGs and will be a major contributor to the sustained replication of HIV in a person.
Materials and methods
Cell culture preparation
Human embryonic kidney 293T cells (HEK 293T) and CEM cells (T lymphoblast) were obtained from the American Type Culture Collection (ATCC). HEK 293T cells were cultured and maintained in DMEM with 5% FBS and 1% penicillin/streptomycin. Phoenix-Ampho cells (purchased from ATCC) are a derivative of HEK 293T cells engineered to package retroviruses for transduction and were cultured similarly to the base HEK 293T cells.23,24 CEM cells were cultured and maintained in Roswell Park Memorial Institute media with 5% FBS and 1% penicillin/streptomycin. Cells were incubated at 37°C and 5% CO2 in a humid environment. All growth media and supplements were purchased from Gibco.
HIV plasmids
HIV plasmids Vpr, Vif, Nef were generously donated by WC Greene (University of California San Francisco) and Vpu was donated by K Strebel (National Institutes of Health, National Institute of Allergy and Infectious Diseases). FLAG-STAT1 and GFP-IRF9 were purchased from Addgene. FLAG-Tyk2 and HA-Jak1 were purchased from Origene. Gag components including matrix, capsid, nucleocapsid, p6 and retropepsin were also purchased from Origene. Vpu, Nef and Vif plasmids with a GFP tag were produced by PCR and introduced into the pBMN plasmid.
Luciferase reporter assay
Luciferase reporter assay was performed to measure the ISRE-containing promoter activity. HEK 293T cells were transfected in 24-well dishes as sets of triplicates. In brief, triplicates were transfected with X-tremeGENE™ 9 (Roche) at a ratio of 2:3 (transfection reagent: DNA). Each triplicate was transfected with 1250 ng of total plasmids with a mix of 200 ng of pGL3-ISRE luciferase reporter, 50 ng of pSV40-Renilla luciferase reporter and 1000 ng of expression plasmid. Transfection complex was incubated in 75 µl of DMEM with X-tremeGENE™ 9 per triplicate for 20 min. HEK 293T cells were plated at 1 × 105 cells total per well. To each well 20 µl of transfection complex was added. Transfections were incubated for 24 h. The next day, transfections were stimulated with 1000 U of IFN-α for 6 h. Then, cells were lysed with passive lysis buffer from the Dual-Luciferase Reporter Assay kit (Promega). Luciferase readings were obtained by the GloMax®-Multi Detection System and analyzed using Microsoft Excel.
Vpu and Nef co-transfection
Co-transfection was performed to measure the expressions of IFN-α pathway components. HEK 293T cells were co-transfected with 1000 ng of control plasmid or an expression plasmid for Vpu or Nef plasmid and 1000 ng of one of the following plasmids: FLAG-Tyk2, HA-Jak1, FLAG-STAT1 or GFP-IRF9 in each well of a six-well plate. Transfection reagent, X-tremeGENE™ HP (Roche) was used at a ratio of 1:1 (extreme gene HP: DNA plasmids). Each transfection complex was incubated in 100 µl of DMEM/X-tremeGENE™ for 20 min. After incubation, 100 µl of transfection complex was added to each well, which had been plated with HEK 293T cells at a concentration of 4 × 105 cells/ml (8 × 105 cells total per well). Transfections were incubated for 48 h. Whole-cell lysates were obtained by incubating cells in 1X SDS loading buffer (1X Laemmli buffer (Bio-Rad) supplemented with 10% DTT). Samples were analyzed by Western blot analysis.
Stable CEM cell line establishment
Stable cell lines were established to measure the endogenous level of IFNAR-signaling components and their phosphorylated states. Phoenix-Ampho was used to package retroviruses with control, Vpu, Nef or Vif (as a control) that could be used to transduce CEM cells. Phoenix-Ampho cells were transfected with 2000 ng of plasmids using X-tremeGENE™ 9 (Roche) (2:3 ratio). Transfected cells were allowed to package viruses for 48 h. Retrovirus was harvested, and the supernatants were filtered through 0.22 µm syringes that had 0.4 µg of polybrene added to them. Retroviruses were added to CEM cells that were plated at 3 × 105 cells per well. CEM cells were spinoculated at 800 g for 2 h in a room temperature (25°C) centrifuge to allow viral attachment. The media uses for packaging was removed and 2 ml of CEM media was added to each well. The cells were incubated for 2 d to allow for productive infection then neomycin was added to allow for selection for about 2 wk. Successfully transduced cells should be able to express EGFP as the control plasmid was GFP expressing and all of the HIV gene Open Reading Frames (ORFs) were fused in frame with Enhanced Green Fluorescent Protein (EGFP). The expression of transduced genes was monitored by GFP expression in the cell population by using flow cytometry. For cells that were selected for vesicular stomatitis virus (VSV)-GFP infections, the transduced ORFs did not contain GFP and thus were monitored by cell recovery after selection. 25
IFN-α stimulation of transduced CEM cells experiment
Stably transduced CEM cells were plated at 3 × 105 cells per well in a 24-well plate and stimulated with IFN-α for 4 h. Whole-cell lysates were obtained by incubating cells in 1X SDS-loading buffer (1X Laemmli buffer (Bio-Rad) supplemented with 10% DTT). Samples were analyzed by Western blot analysis.
Western blot
Whole-cell lysates were passed through a 21-Gauge needle three times to shear cellular DNA and membranes, then boiled for 5 min to denature protein. Samples were clarified by spinning at 14,200 g for 10 min in a microcentrifuge. Then 30 µl of each sample were run in a pre-made TGX gel (Bio-Rad) for 30 min at 200 mV in 1X Tris-SDS running buffer (Bio-Rad). Proteins were transferred using the Trans-Blot Turbo Transfer system (Bio-Rad). Each membrane was blocked in 5% milk TBST for 1 h at room temperature on a shaker. Specific primary Abs were added and incubated overnight in 5% milk TBST at 4°C with agitation. The Abs used were anti-FLAG M2 Ab (Sigma) (1:1000), anti-GFP (FL) Ab (Santa Cruz Biotech) (1:500), (1:500), anti-HA Ab (Sigma) (1:10,000), anti-STAT1 Ab (Cell Signaling) (1:3000), anti-pSTAT1 Ab (Cell Signaling) (1:1000), anti-Tyk2 (Cell Signaling) (1:1000), anti-actin (Cell Signaling) (1:10,000) and anti-tubulin (Cell Signaling) (1:10,000). For pSTAT1 Ab, 5% BSA TBST was used instead of 5% milk TBST for membrane blocking and primary Ab incubation. Membranes were washed the next day three times (5 min each time) with 5% milk TBST. Membranes were incubated in secondary Ab, either goat anti-mouse (Santa Cruz Biotech) (1:3000) or goat anti-rabbit (Santa Cruz Biotech) (1:3000) for 1 h. Then, membranes were washed three times with 5% milk TBST (15 min each time) and three times with TBST (5 min each time) at room temperature. All the washes were done on a Belly Dancer shaker. Membranes were incubated in Luminol reagent (Santa Cruz Biotechnology) and proteins were detected using digital light detection using a Gel Doc XR+ System and analyzed by Image Lab (BioRad).
Note on values graphed in figures
In an effort to show genuine quantitative data, we do not show fold induction for any luciferase experiment but instead show the actual values obtained in an experiment normalized to the internal standard. We feel this gives a more genuine representation of data and that the data are not masked through the use of fold-induction values, especially in reporter assays. Consequently, the values in different experiments may vary but the interpretation remains the same and is more consistent with the biology of each individual experiment. Relative levels are added above the real data graphed.
Results
Vpu and Nef reduce IFN-α activation of ISRE promoter activity
To begin investigating if HIV modulates IFN-α signaling, we first sought to identify which of the HIV proteins are involved in controlling IFN-α signaling. IFN-α, induced during viral infection, activates the transcription factor ISGF3, which binds to the promoter element ISRE to start transcribing ISGs. 26 Therefore, we measured the activity of the ISRE promoter in the presence of HIV proteins using luciferase assays. We screened all the possible HIV protein products by co-transfecting HEK 293T cells with plasmids individually expressing each of the HIV proteins and the ISRE luciferase plasmid. We also included a positive control, protein inhibitor of activated STAT, which is known to inhibit JAK/STAT signaling. 27 The cells were transfected with the corresponding plasmids for 24 h and then stimulated for 6 h with 1000 U of IFN-α. Cell lysates were obtained and the luminescence levels were measured (Figure 1). Data obtained were normalized to the levels of Renilla, which was used as an internal control.
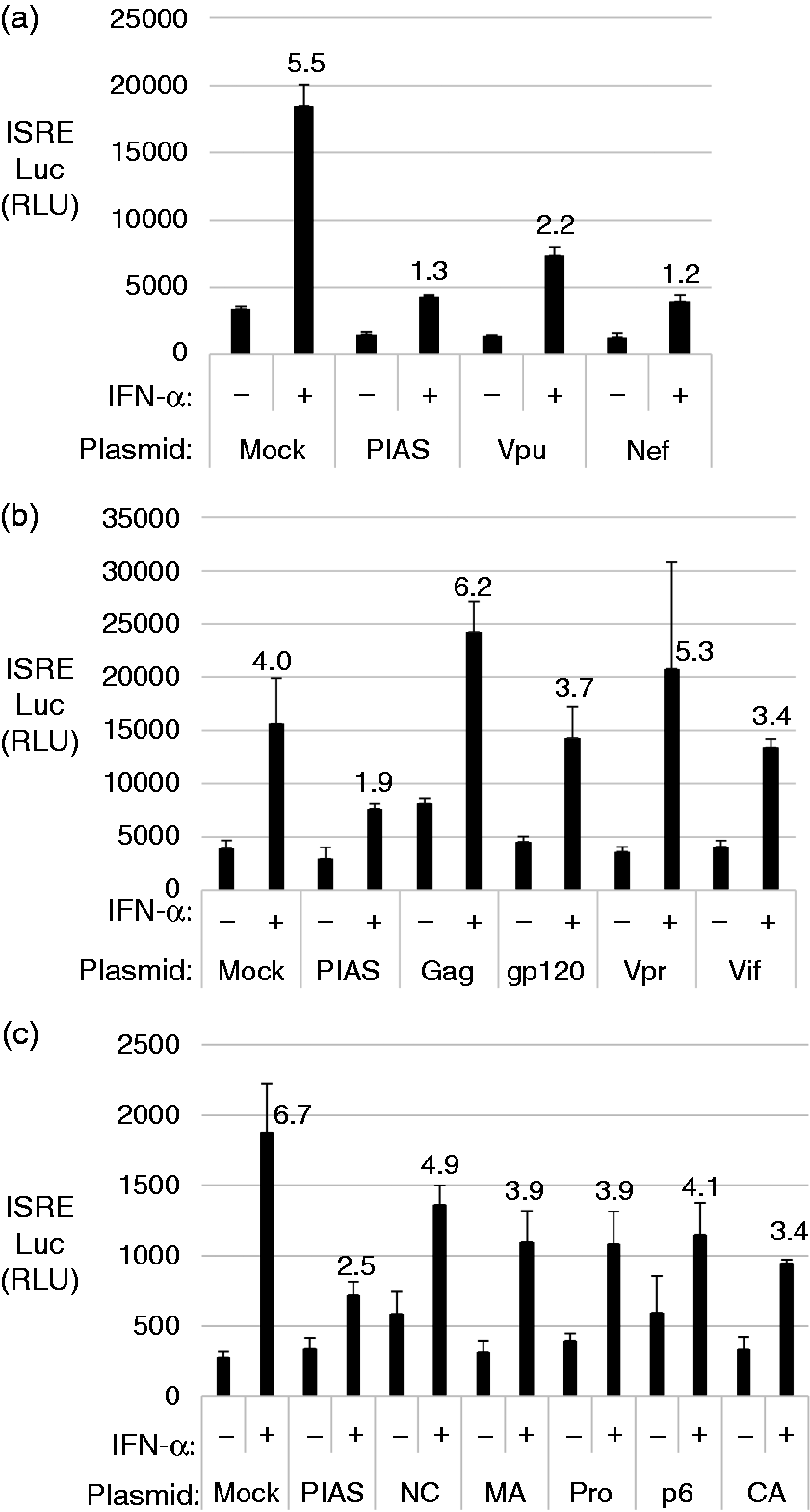
HIV Open Reading Frames (ORFs) block IFN-α-induced IFN-stimulated response element (ISRE) activation. HEK 293T cells were transfected with plasmids expressing individual HIV proteins for 24 h and then stimulated with 1000 U of IFN-α for 6 h. ISRE reporter activity was measured and graphed in relative luciferase units normalized to the internal control Renilla. Graphed data represent the average of a triplicate done on the same day and each experiment was repeated three times. The graph displays data from transfection with (a) Vpu and Nef, (b) gag, gp120, Vpr, Vif, (c) nucleocapsid (Nc), matrix (Ma), protease (Pro), p6 and capsid (Ca). Fold changes after IFN-α stimulation are noted above the respective bars.
Upon transfection of HIV plasmids, compared to the control transfection, we found that IFN-α-induced ISRE-luciferase signal was lower in the Vpu and Nef transfected cells (Figure 1a). ISRE promoter activity was significantly reduced in the presence of Vpu and Nef compared to transfection with the other HIV proteins including gp120, Vpr, Vif, protease (retropepsin) and the Gag components: nucleocapsid, capsid, matrix, p6 (Figure 1b and 1c).
These experiments were repeated in a dose-dependent manner with increasing amounts of Vpu and Nef. As seen in Figure 2a for Vpu and 2b for Nef, the levels of the ISRE activity decreased as Vpu and Nef increased. Therefore, the ISRE promoter activity was inhibited specifically by Vpu and Nef but not by other HIV components.
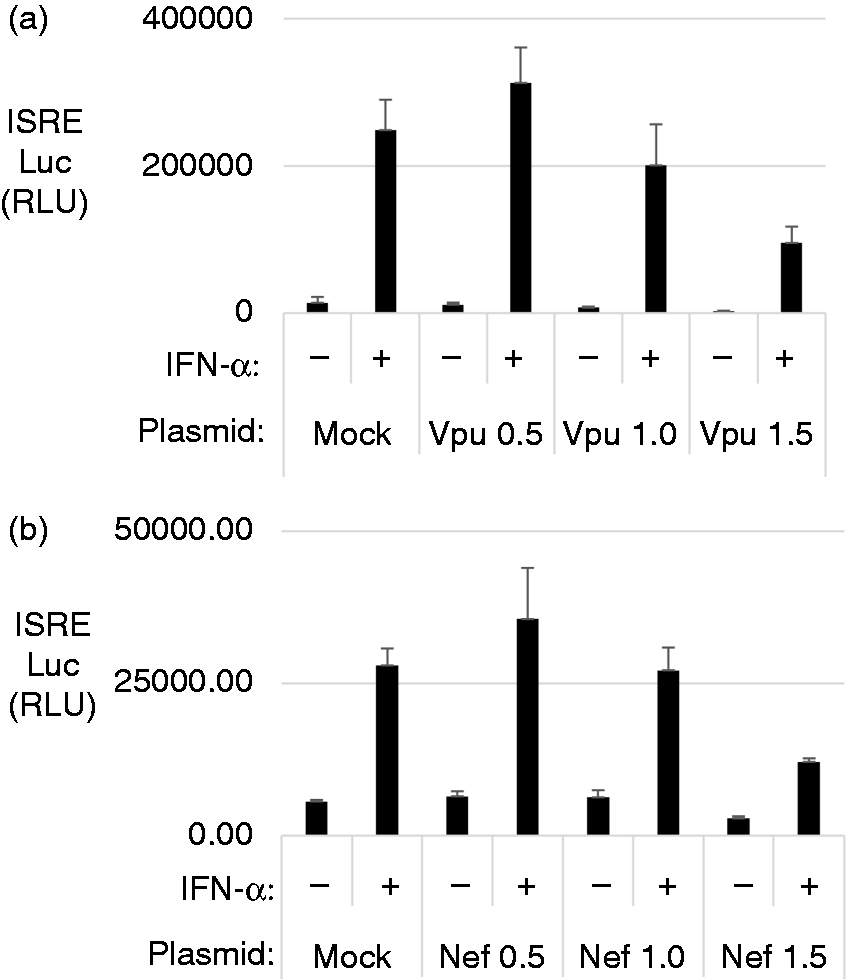
Vpu and Nef specifically block IFN-stimulated response element (ISRE) activation. (a and b) HEK 293T cells were transfected with plasmids expressing individual HIV proteins for 24 h and then stimulated with 1000 U of IFN-α for 6 h. ISRE reporter activity was measured and graphed in relative luciferase units normalized to the internal control Renilla. Graphed data represent the average of a triplicate done on the same day and each experiment was repeated three times. Transfections for the luciferase assay were repeated in a dose-dependent manner with 0.5 µg, 1.0 µg and 1.5 µg of Vpu in (a) or Nef in (b). Graphed data represent the average of a triplicate done on the same day and each experiment was repeated three times.
Jak1 and Tyk2 protein expressions are unaffected by Vpu and Nef
Because ISRE promoter activity was inhibited by Vpu and Nef, we hypothesized that Vpu and Nef must interact with a cellular component somewhere in the IFN-α signaling pathway. To determine at which step of the signaling pathway Vpu and Nef have an effect, we first looked at the stability of components of the JAK/STAT pathway and assessed the level of expression of the two kinases, Jak1 and Tyk2 in the presence of Vpu and Nef. Jak1 and Tyk2 kinases are activated following the binding of IFN-α to the IFNAR receptor. 28 Jak1 and Tyk2 activate intermediate proteins, which include STAT1 and STAT2 downstream.
HEK 293T cells were co-transfected with HA-tagged Jak1 or FLAG-tagged Tyk2 plasmids along with Vpu or Nef plasmids. The protein levels of Jak1 and Tyk2 were not significantly reduced by Vpu or Nef (Figure 3a and b). Therefore, Vpu and Nef do not affect the expression of Jak1 and Tyk2 even though ISRE activity was inhibited by Vpu and Nef.
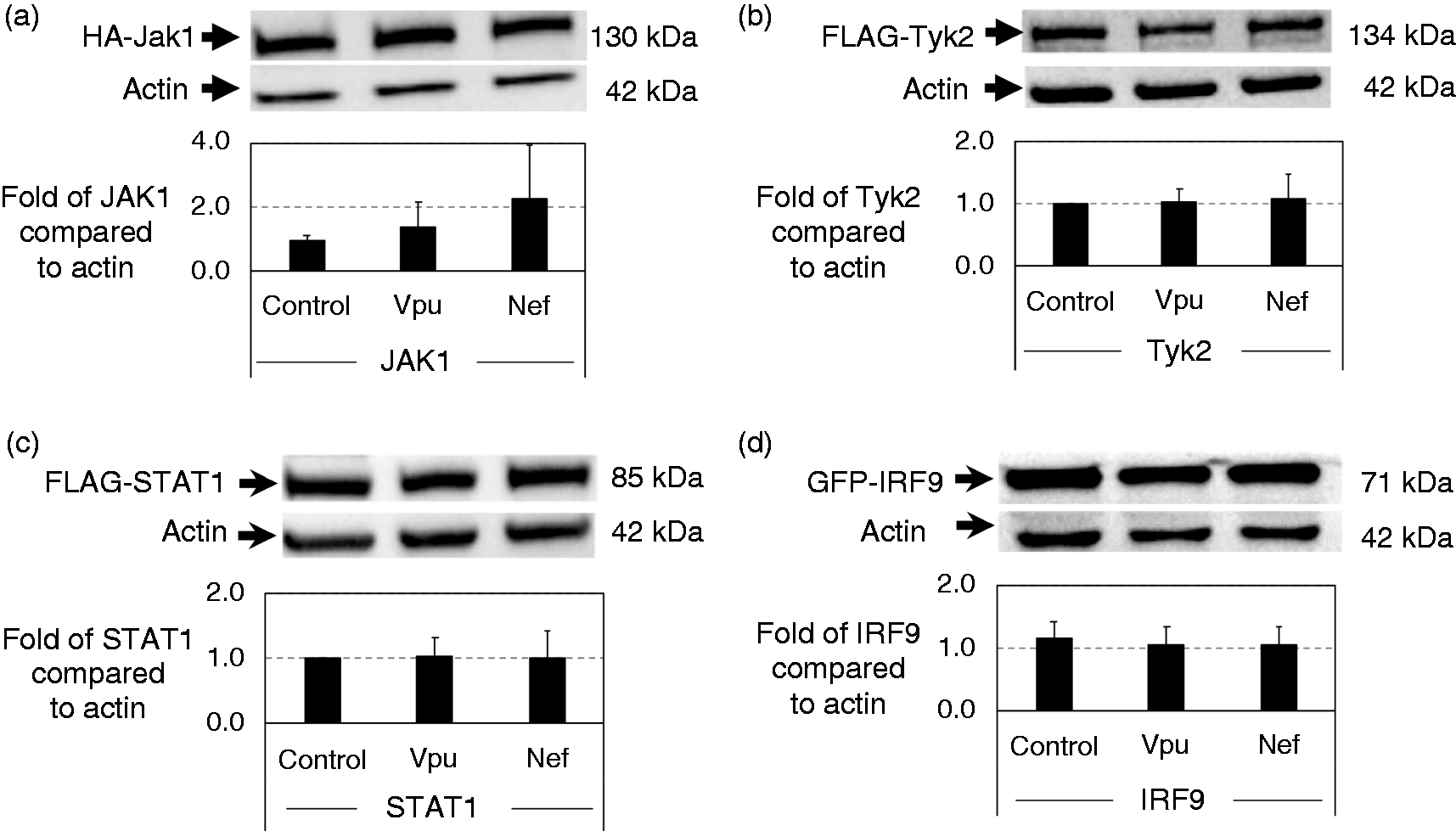
Components of IFN-α/β receptor signaling are stable in the presence of Vpu or Nef. HEK 293T cells were co-transfected with HIV plasmids (Vpu or Nef) and (a) HA-tagged Jak1, (b) FLAG-tagged Tyk2, (c) FLAG-tagged STAT1 or (d) GFP-tagged IRF9. After 48 h of incubation, protein lysates were obtained by re-suspending in 1X SDS loading buffer for Western blot analysis using actin as a loading control. After experimental setup and establishment as well as Ab titrations, each Western blot was additionally repeated an additional three times as displayed. The relative protein expressions relative to actin levels for four replicates of the Western blot were graphed along with standard deviation.
STAT1 and IRF9 are unaffected by Vpu and Nef
Next, we looked at the two proteins downstream of the JAK/STAT pathway. STAT1 is phosphorylated by activated Jak1 kinase. 29 When phosphorylated, STAT1 is able to dimerize with phosphorylated STAT2. This heterodimer can recruit IRF9 to form a transcription factor complex called ISGF3 that can then enter the nucleus to activate ISRE-containing promoters. Here, we examined the expression levels of STAT1 and IFR9 in the presence of Vpu and Nef.
HEK 293T cells were co-transfected similarly to the experiment above with FLAG-tagged STAT1 or GFP-tagged IRF9 plasmids along with Vpu or Nef plasmids. Whole-cell lysates were obtained following 48 h of incubation. Surprisingly, both STAT1 and IRF9 were unaffected by Vpu and Nef as seen in Figure 3c and d. Thus, although Vpu and Nef reduce IFN-α activation of ISRE, they do not affect the stability of the kinases Jak1 or Tyk2, or two key components of the ISFG3 activator, STAT1 or IRF9.
Phosphorylation of STAT1 is inhibited by Vpu and Nef
So far, expression of the proteins in the IFN-α pathway was shown to be unaffected by Vpu and Nef. Because ISRE activation is being blocked, we hypothesized that perhaps the phosphorylation of STAT1 is affected by Vpu or Nef. To understand the phosphorylation activity, we looked at the endogenous STAT1 and its phosphorylation state. We engineered CD4+ T cell lines that stably express HIV proteins by transducing CEM cells using retrovirus produced from Phoenix-Ampho cells. We used Vpu, Nef and a control GFP plasmid to obtain three different cell lines. Flow cytometry was performed to monitor uniform gene expression by looking at GFP-expression in the cell population (Figure 4f). Stably transduced CEM cells were plated and stimulated with IFN-α for 2 and 4 h. Protein levels for Tyk2, STAT1 or phosphorylated STAT1 were monitored by Western blot.
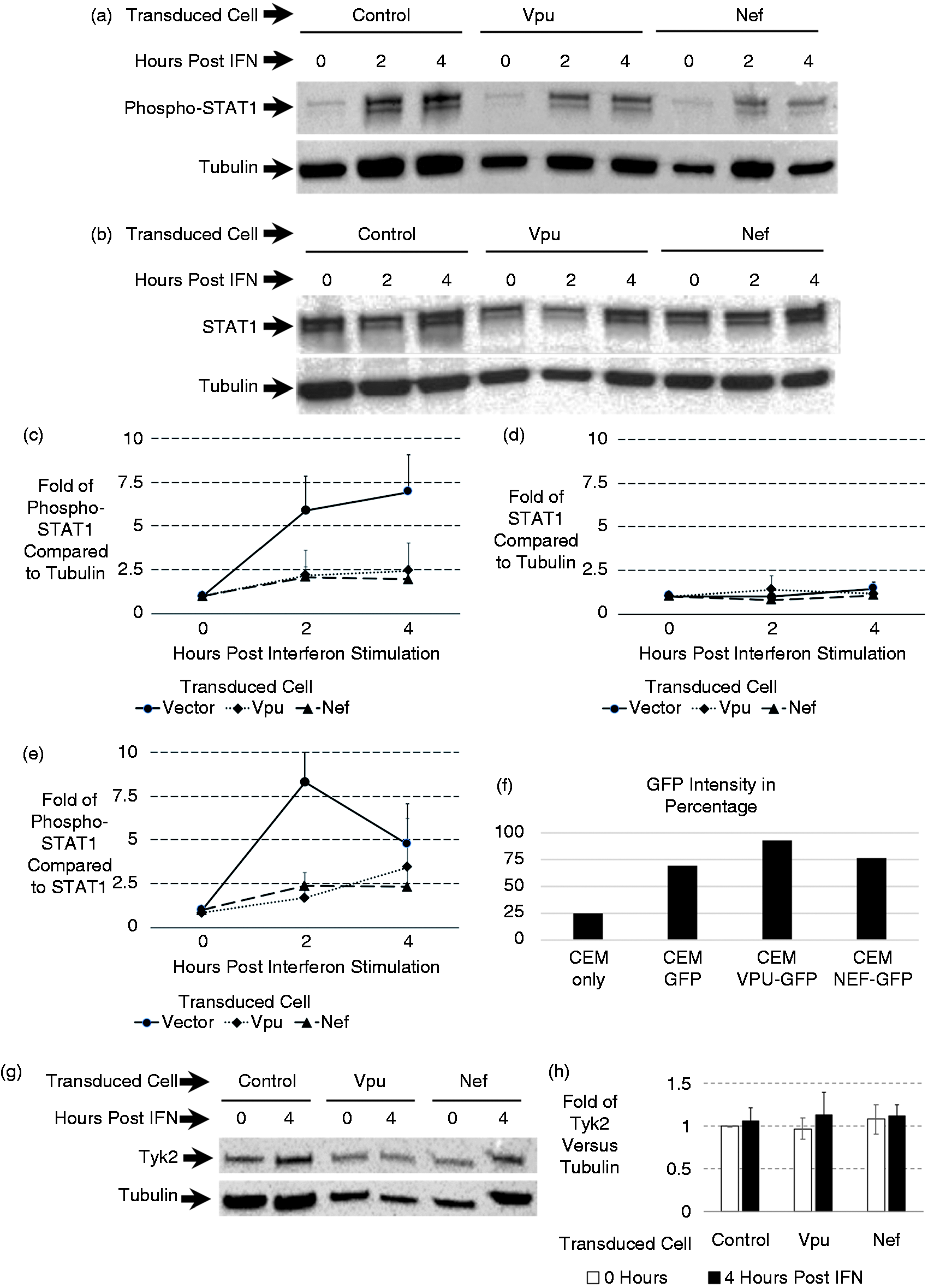
Vpu and Nef block IFN-α stimulated STAT1 phosphorylation. Stable CEM cell lines expressing Vpu and Nef were established. Transduced cells were selected between 10–14 d and monitored by flow cytometry based on the presence of GFP. Cells were plated and stimulated with 1000 U of IFN-α for 2 or 4 h. Cell lysates were obtained in SDS loading buffer and analyzed by Western blotting for (a) phosphorylated STAT1 or (b) total STAT1 along with tubulin for each. After the experimental setup and establishment as well as Ab titrations, each Western blot was additionally repeated an additional three times as displayed. The relative protein expressions relative to actin levels for four replicates of the Western blot were graphed along with standard deviation. Relative fold of protein levels for phosphorylated STAT1 were graphed in (c) and for total STAT1 were graphed in (d). Levels of phosphorylated STAT1 compared to the corresponding STAT1 were graphed in (e). (f) Displays GFP levels of the transduced population to show that all cell lines were similar in terms of transduction and stable expression of genes. (g) Total Tyk2 levels at 0 and 4 h after IFN-a stimulation. The relative protein expressions relative to actin levels for four replicates of the Western blot were graphed along with standard deviation. Relative fold of protein levels for Tyk2 were graphed in (h).
As shown in Figure 4, there is a reduction of phosphorylated STAT1 after IFN-α stimulation in the Vpu- and Nef-expressing cells (Figure 4a). Nef seemed to have more impact in decreasing the phosphorylation of STAT1 than Vpu. Meanwhile, GFP did not have any effect on the phosphorylation state of STAT1. The data were consistent with the luciferase assay result where Nef also seemed to have stronger inhibition of ISRE promoter activation compared to Vpu. In another set of experiments when blotting for STAT1 and Tyk2, the expression levels of these proteins were not affected by Vpu or Nef as seen in the results (Figure 4b and 4g). Fold of protein levels was plotted for four replicates of each graph (Figure 4). These data were also consistent with the results in Figure 3, where the expression levels of Tyk2 and STAT1 were not affected in the transient transfection experiment.
Vpu increases VSV replication in the presence of exogenous IFN-α
To better understand the physiological impact of targeting STAT1 phosphorylation as a mechanism of diminishing ISRE and ISG activation, we utilized an IFN-induced antiviral efficacy assay. If Vpu disrupts the expression of ISGs in the stable cell lines, we expect to see a lack of viral protection in these cells. We designed an experiment to understand the infection rate of VSV-GFP in the presence of HIV proteins after those cells were stimulated with IFN-α. The stable cells were stimulated with IFN-α for 4 h followed by VSV-GFP infection for 24 h. The cells were collected and analyzed by flow cytometry looking at the level of GFP.
VSV-GFP expresses GFP if infection is allowed to proceed (Figure 5a vs 5 b). However, the addition of IFN-α leads to a decreased level of GFP (Figure 5b vs 5 c). As expected, comparing stable Vpu cells to stable Vif and CEM cells, GFP level was increased in Vpu stable cells relatively (Figure 5e vs 5 h). Therefore, the rate of VSV replication was higher when there was Vpu because the antiviral activities were probably reduced. Additionally, when treated with IFN-α to activate the antiviral state, the control CEM cells and those stably expressing Vif cells were rescued as shown by decreased VSV-GFP levels (Figure 5c and 5f). Meanwhile, in the presence of Vpu, the IFN-α-induced rescue effect did not take place. There was no difference with and without treatment of IFN-α as Vpu seems to disrupt the function of IFN-α interfering with VSV-GFP replication (Figure 5h vs 5 i). In short, Vpu decreases the ability of IFN-α to induce an efficacious antiviral state and allowed for the increased replication and spread of VSV. Representative fluorescent pictures of the cells analyzed in Figure 5a–5i are shown in Figure 5j.
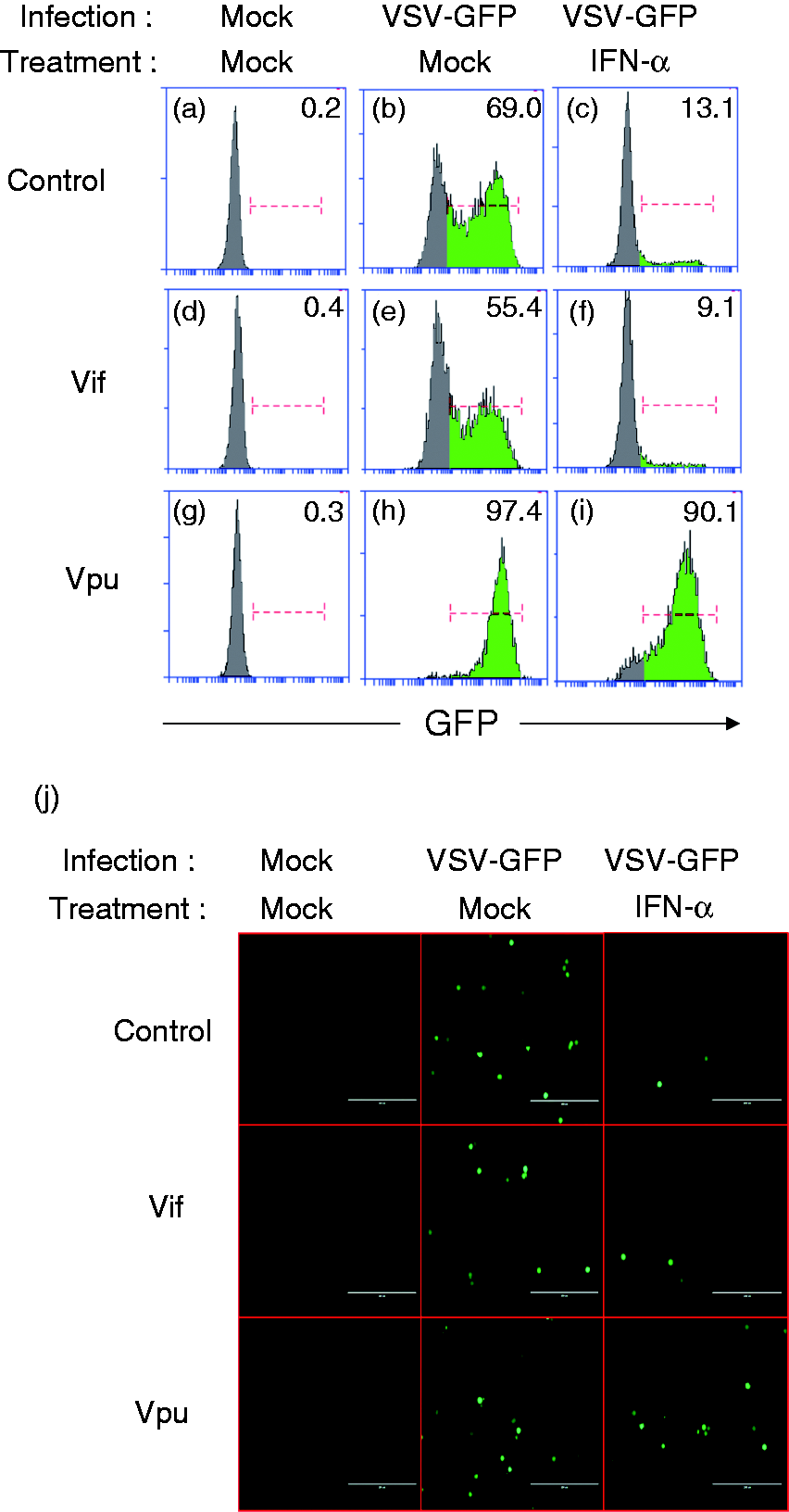
Vpu blocks IFN-α stimulated establishment of an antiviral state. (a–i) Stable CEM cell lines expressing Vpu or Vif were established. Cells were plated and stimulated with 1000 U of IFN-α for 6 h and infected with a GFP expressing vesicular stomatitis virus. Cells were monitored by flow cytometry based on the presence of GFP. The percentage of GFP-positive cells is indicated at the top right corner. (j) Fluorescent pictures of the cells were taken, and representative pictures are displayed. The scale bar is at 200 µm.
Discussion
Upon the recognition of HIV via RLRs or TLRs, IFN-α is produced and released to activate the JAK/STAT pathway to signal for the expression of ISGs. ISGs have multiple functions in combatting viral infections. However, during HIV infection, HIV encodes multiple proteins that target this critical system. HIV accessory proteins including Vpu and Nef are vital to promote increasing HIV replication. The ability of Vpu and Nef to interact with multiple signaling proteins and direct these components toward degradative pathways is well documented. 30 Our group has shown that Vpu and Nef are able to block this pathway by degrading IPS-1, a protein in the RLR signaling cascade. 14 By degrading IPS-1, the release of IFN-α is reduced from the infected cells. Meanwhile, viral-sensing immune cells such as pDCs and macrophages induce the production of IFN-α through TLR-dependent pathways. Furthermore, pDCs can produce IFN-α 1000-fold higher than any other cell types in the immune system. The systematic induction of IFN-α by pDCs and macrophages can still contribute to the global protection against HIV through the IFN-α pathways.7,31 Our current studies show that even with IFN-α released into the local microenvironments in response to HIV infection, HIV is still able to block IFN-α-dependent antiviral activities.
First, we show that out of the entire HIV genome, activation of the ISRE promoter by IFN-α stimulation was reduced only in the presence of Vpu and Nef. Furthermore, this effect had a dose dependency as transfections of lower levels of Vpu or Nef had less effect on IFN-α-induced ISRE activation. Therefore, the levels of Vpu and Nef expression must reach a certain threshold to effectively inhibit the promoter activity, presumably during active lytic replication of HIV. Because the ISRE promoter activity is inhibited in the presence of Vpu or Nef, one could speculate that somewhere in the pathway, signaling intermediates were targeted. However, we show the expressions of Tyk2, Jak1, STAT1 as well as IRF9 were targeted by neither Vpu nor Nef. We then find that Vpu and Nef reduce the phosphorylation of STAT1.
Phosphorylation of STAT1 is one of the crucial steps in activating Type I IFN response. 32 HIV is not the only virus that can down-regulate the JAK/STAT pathway. One study showed that Kaposi's sarcoma-associated herpesvirus inhibits IFN-α signaling by a viral gene product, RIF, which can form complexes with Jak1, Tyk2, STAT2 and IFNAR subunits. 33 RIF can relocate STAT2 to IFNAR1 in the absence of IFN-α, and it also reduces the activity of Tyk2 and Jak1 kinases. Another study also showed an inhibition of IFN-α signaling by hepatitis C virus (HCV). 34 HCV can up-regulate a microRNA, mir-373, which controls the expression of Jak1 and IRF9 by forming complexes with mRNAs and preventing them from being translated. Both these viruses can disrupt IFN-α signaling, which leads to reduced ISG expression. HIV is also able to inhibit this signaling pathway via inhibition of pSTAT1 by Vpu or Nef. This result might indicate that HIV can still replicate and bypass the innate immune protection in the presence of IFN-α. IFN-α release is important during early phases of HIV infection to prevent the spread of the virus to healthy cells as IFN-α is activated by the JAK/STAT signaling pathway to provide multiple defensive mechanisms against the virus. However, Vpu and Nef disrupt the JAK/STAT pathway induced by IFN-α and down-regulate ISG expressions in HIV-infected cells (Figure 6). Thus, infected cells are not able to control viral production effectively even with the presence of IFN-α in the microenvironment. We also observed a reduced antiviral effect of IFN-α with an antiviral efficacy assay using infection of VSV-GFP in a Vpu stable cell line. This observation confirmed that the antiviral protection by ISGs is less effective because Vpu blocks the signaling pathway that activates ISG expression. In addition, the local IFN-α induction is already reduced via degradation of IPS-1 by Vpu and Nef. Therefore, Vpu and Nef block the IFN-α signaling pathway at two steps, the release of IFN-α via signaling from the RLR-dependent pathway and the activation of the JAK/STAT pathway downstream by IFN-α. It is important to note that others have shown that Vif may also target the IFN/JAK/STAT pathway, although our data in Figure 5 would seem to encourage further research into these findings. 35 A limitation of these studies is that we have not determined the impact of Vpu and Nef on IFN-α during HIV infection. Although VSV serves as a well-studied monitor for IFN-α function in cells, it will be important to extend our work to primary infection of HIV. An important note is that without Vpu and Nef, HIV replication is highly diminished and may confound those studies.
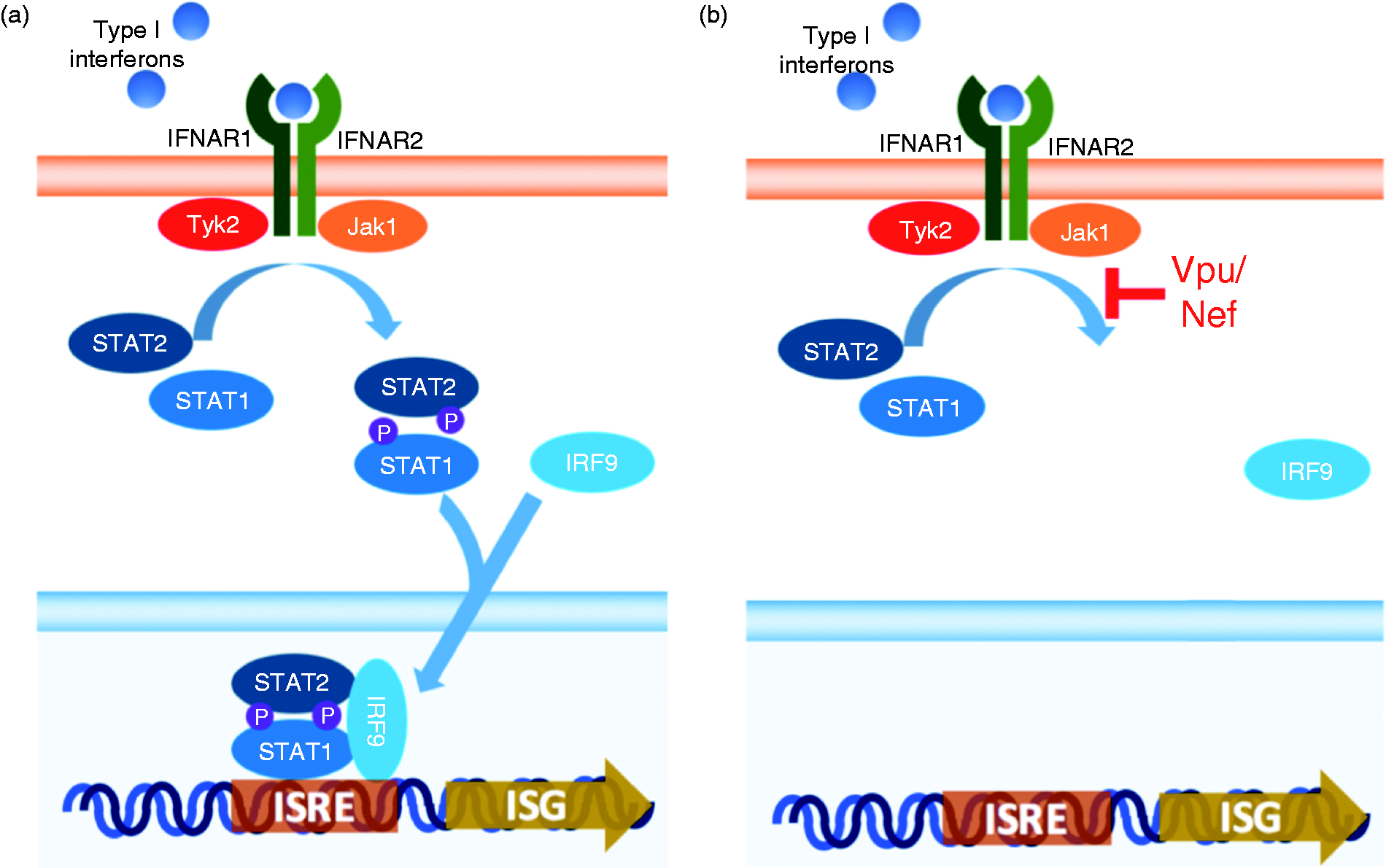
Proposed mechanism of Vpu/Nef block to antiviral state establishment. (a) IFN-α leads to establishment of the antiviral state via induction of IFN-α/β receptor signaling that ultimately leads to up-regulation of ISGs. (b) Vpu and Nef block phosphorylation of STAT1, which could lead to a dampening of IFN-α induction of an antiviral state.
Short-term treatment of HIV using IFN has been shown to lower HIV viral loads. 36 However, long-term IFN treatment might be more harmful and less beneficial to patients’ immunity because IFN can contribute to continuous immune activation and new viral establishment from latent reservoirs. 37 Over time, exogenous IFN will deplete CD4+ T cells. In addition, IFN is not a desirable treatment as it introduces many side effects as seen in hepatitis C patients.38,39 Our data indicate that exogenous IFN-α responses are blocked by Vpu and Nef. This further implies that IFN is not effective as a drug for HIV. Meanwhile, by disrupting the functions of Vpu and Nef, the natural IFN-α responses might be restored. This approach could avoid the risks of side effects and damage to the immune system by letting innate immunity combat HIV naturally.
Further investigation is needed to understand the mechanism of pSTAT1 inhibition by Vpu and Nef. One possibility might be the kinase activity of Tyk2 or Jak1 is blocked by HIV but not necessarily through modulation of the expression of Tyk2 or Jak1, leading to the inability of these kinases to phosphorylate STAT1. Another possibility might be the IFNAR is unable to activate Tyk2 or Jak1, thus preventing further activation of downstream signaling though their gene expressions are unaltered. If these mechanisms are understood, strategies on how to block Vpu and Nef can be examined and studied to suppress HIV replication.
In conclusion, HIV seems to exhibit multiple blocks of innate immunity as seen in the down-regulation of APOBEC3G by Vif, degradation of IPS-1 and inhibition of pSTAT1 by Vpu and Nef. Indeed, several other groups have shown that HIV proteins can disrupt interferon induction through a variety of means.15,40,41 Particularly, multiple blocking points throughout the IFN-α pathways by Vpu and Nef underscore the importance of proper IFN-α signaling in controlling HIV replication. This global block explains why exogenous IFN-α treatment might not work on HIV clinically, as the blockade to the JAK/STAT signaling will stop the activation of all ISGs. Due to excessive IFN-α released in the serum of patients infected by HIV, blocking Vpu or Nef function may be sufficient to allow for a patient’s own innate immunity to stop HIV replication leading to better efficacy of therapy and treatment.
Footnotes
Acknowledgements
NVN and JTT were supported by the Western University of Health Sciences College of Pharmacy through the Masters in Science in Pharmaceutical Sciences programs. DJS was partially supported by National Institutes of Health 1R15AI138847.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.