
Abstract
Elastase released by neutrophils is critical for eliminating Gram-negative bacteria. Ca2+ influx plays a key role in elastase release and bacterial clearance in neutrophils. Transient receptor potential melastatin 2 (TRPM2) is a Ca2+-permeable cation channel highly expressed in neutrophils. Here, we explore the role and possible mechanism of TRPM2 in bacterial clearance in TRPM2 knockout (TRPM2-KO) mice neutrophils. After exposure to Escherichia coli, TRPM2–KO bone marrow neutrophils (BMNs) had increased bacterial burden and decreased elastase release. The same was observed for septic TRPM2-KO mice which also had decreased survival rate. After stimulation with chemotactic peptide N-formyl-methionyl-leucyl-phenylalanine (fMLP), elastase release was lower in TRPM2-KO BMNs than in wild type (WT) BMNs. Pre-treatment of WT BMNs with p38 MAPK inhibitor reduced fMLP-induced elastase release. Compared with WT BMNs, TRPM2-KO BMNs had decreased p38 MAPK phosphorylation after fMLP stimulation. Removal of extracellular Ca2+ reduced fMLP-induced p38 MAPK phosphorylation and elastase release. The concentration of intracellular Ca2+ decreased in TRPM2-KO BMNs compared with WT BMNs after fMLP treatment. Hence, TRPM2 plays an important role in bacterial clearance in neutrophils, possibly by regulating elastase release. TRPM2-mediated Ca2+ influx regulates elastase release partially via p38 MAPK phosphorylation in neutrophils.
Introduction
Neutrophils are critical for the first-line host innate immune defense and resistance to microbial infection.1,2 N-Formyl-methionyl-leucyl-phenylalanine (fMLP), a peptide from the Gram-negative bacterial cell wall, is a potent agonist of neutrophil activation implicated in innate host immunity. 2 fMLP triggers the phosphorylation of intracellular tyrosine kinase and downstream p38 MAPK, leading to neutrophil degranulation.3,4 Among the proteinases released from azurophil granules, elastase is a potent serine proteinase which plays a key role in eliminating Gram-negative bacteria in neutrophils.5,6
Transient receptor potential melastatin 2 (TRPM2) is a non-selective Ca2+-permeable cation channel which is highly expressed in immune cells including neutrophils and macrophages.7–12 TRPM2 is potently activated by intracellular ADP-ribose (ADPR) through binding to the unique NUDT9 homology domain in its distal C-terminus. 8 TRPM2 channels can also be activated by various factors such as hydrogen peroxide (H2O2), intracellular Ca2+, cyclic ADPR, and nicotinic acid adenine dinucleotide.7–9 In response to fMLP, basal level of ADPR can sufficiently activate TRPM2 through mobilization of intracellular calcium from the IP3 receptor when intracellular Ca2+ is elevated.13–15 Moreover, fMLP recruits the nicotinamide adenine dinucleotide phosphate-oxidase to the membrane 16 and strongly stimulates H2O2 release.17,18 H2O2 release can also contribute to activation of TRPM2. 19
The receptor for fMLP is a classical G protein-coupled receptor that triggers a biphasic calcium transient characterized by an early peak followed by a plateau phase in neutrophils stimulated with fMLP. The early peak is associated with mobilization of intracellular calcium from the IP3 receptor. This leads to a sustained calcium influx through membrane calcium channels.20–22 Studies have shown that TRPM2 seems to represent a critical membrane Ca2+ influx channel in neutrophils in response to fMLP. TRPM2-knockout (KO) neutrophils showed impaired calcium influx and migration in response to fMLP.9,23
Numerous studies suggest an important role of macrophage TRPM2 in regulating inflammation24–26 and bacterial clearance.27–29 Our previous study demonstrated the protective role of macrophage TRPM2 in controlling bacterial clearance during polymicrobial sepsis, possibly by regulating heme oxygenase-1 expression. 28 Another study further explored the detailed mechanism of macrophage TRPM2 in bacterial killing and found that TRPM2-mediated Ca2+ influx plays an important role in bacterial clearance through promoting phagosome maturation in Escherichia coli sepsis. 29 Ca2+ influx plays a key role in azurophil granule release and bacterial clearance in neutrophils.30,31 One previous study found that TRPM2 is involved in lysophosphatidylcholine (LPC) enhancement of neutrophil bactericidal activity by increasing azurophil granule–phagosome fusion/elastase release. 31 However, the role and the possible mechanism of TRPM2 in bacterial clearance in neutrophils has still not been fully elucidated. Herein, we hypothesized that TRPM2 is required for bacterial clearance in neutrophils by regulating elastase release.
In this study, we first investigated whether elastase release and bacterial clearance were decreased in E. coli-treated TRPM2-KO neutrophils. We next investigated whether elastase release and bacterial clearance were reduced in the peritoneal cavity in cecal ligation and puncture (CLP)-induced septic TRPM2–KO mice. Finally, we explored how TRPM2 affected elastase release in neutrophils.
Materials and methods
Mice and sepsis model
Male wild type (WT) mice (C57BL/6) were purchased from Zhejiang Province Experimental Animal Center. TRPM2-KO mice were generated by Yamamoto et al. 23 and had been backcrossed more than 12 generations onto the C57BL/6 background. Male mice aged 8–12 wk (20–30 g body mass) were used in the study. The animal protocols for experiments were approved by the Animal Experimentation Committees at Zhejiang University (Hangzhou, Zhejiang Province, People’s Republic of China) and Wenzhou Medical University (Wenzhou, Zhejiang Province, People’s Republic of China). Polymicrobial sepsis was performed by CLP as previously described. 28 Mice were anesthetized by intraperitoneal administration of 80 mg/kg pentobarbital. The cecum was exposed via a small abdominal midline incision and ligated at midway between the base of cecum and distal pole using a 4-0 silk ligature and then punctured once through both surfaces with a 21-G needle. After extruding a small amount of fecal material, the cecum was re-positioned, and the abdomen incision was closed. Sham-operated mice underwent the similar procedure but without ligation and puncture. All mice were injected with 1 ml of normal saline subcutaneously for fluid resuscitation after surgery. Survival was monitored twice daily for 7 d. Mice were randomized to different experimental groups and investigators were blinded to mouse genetic background and treatment group.
Peritoneal lavage fluid collection
Peritoneal lavage fluid (PLF) was harvested according to our previous study. 28 Briefly, 16 h after sham or CLP surgery, mice were euthanized. After dampening mice with 70% ethanol for 1 min, half of the abdominal wall was exposed and a 25-gauge needle was carefully inserted into the peritoneal space, avoiding injury to the intestines. The needle was fixed with a vascular clamp. Two separate 3 ml volumes of PBS were injected into the peritoneal space. PLF was harvested by gentle massage of the abdomen for 10 s.
Isolation of mouse bone marrow neutrophils
Mouse bone marrow neutrophils (BMNs) were collected from femurs and tibias of WT or TRPM2-KO mice as described previously. 32 Briefly, bone marrow progenitors were flushed from the bones and were suspended using Ca2+ and Mg2+-free Hanks Balanced Salt Solutions (HBSS) [137 mM NaCl, 0.53 mM KCl, 0.033 mM Na2HPO4, 0.4 mM NaHCO3, 0.044 mM KH2PO4, and 2 mM HEPES (pH7.4)] (Thermo Fisher Scientific, Pittsburgh, PA, USA) containing 10% FBS (Moregate BioTech, Bulimba, QLD, Australia). After eliminating residual erythrocytes with hypotonic lysis, cells were centrifuged and resuspended in 3 ml of 45% Percoll (Amersham Biosciences, Uppsala, Sweden) in Ca2+ and Mg2+-free HBSS containing 10% FBS. Cells were loaded slowly and carefully on top of a Percoll discontinuous density gradient. After centrifuging at 1600 g for 30 min at room temperature (21–25°C), cells at the interface between 81% and 62% and 62% and 55% were collected and diluted in Ca2+- and Mg2+-free HBSS containing 10% FBS. Cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Thermo Fisher Scientific) containing 10% FBS. Cell viability was more than 98% using tryphan blue staining (Thermo Fisher Scientific). Cell purity monitored by Diff Quick staining (Thermo Fisher Scientific) was more than 95%.
Detection of elastase concentration
Some 16 h after sham or CLP surgery, two separate 3 ml volumes of PBS were injected into the peritoneal space. PLF was harvested by gentle massage of the abdomen for 10 s. Next, 4 ml of the lavage fluid was centrifuged at 600 g at 4°C for 5 min and the supernatant was collected for elastase detection. Then 1 ml of BMNs (2 × 106 cells/ml) from WT or TRPM2-KO mice in RPMI 1640 containing 10% FBS was cultured into each well of a six-well tissue culture plate pre-coated with poly-
Bacterial killing by BMNs in vitro
One milliliter of BMNs (2 × 106 cells/ml) was cultured in RPMI 1640 medium containing 10% FBS in a 24-well flat-bottom plate pre-coated with poly-
Western blot assay
Western blot was performed as previously described. 28 BMNs were lysed in 4 × lithium dodecyl sulfate sample buffer (Novex, Carlsbad, CA). Before boiling the lysates at 70°C for 10 min, 10 × sample reducing agent (Novex) was added at a final concentration of 1 ×. Next, 30 µg of protein was separated by SDS-PAGE and transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA). After blocking with 5% nonfat dry milk in Tris-buffered saline with 0.05% Tween-20 (TBST) (Sigma-Aldrich) for 1 h, membranes were incubated overnight in primary Ab solution of phospho-p38 MAPK (1:1000 dilution, rabbit monoclonal anti-phospho-p38 MAPK Ab, Cell Signaling Technology, Inc., Danvers, MA) or p38 MAPK (1:1000 dilution, rabbit monoclonal anti-p38 MAPK Ab, Cell Signaling Technology) on a shaker on ice. The membrane was then incubated with a goat anti-rabbit IgG HRP-conjugated secondary Ab (Amersham Biosciences) for 1 h at room temperature. Protein expression was detected using the enhanced chemiluminescence reagent (Thermo Scientific).
Measurement of intracellular Ca2+
Intracellular Ca2+ concentration was measured using a VARIOSKAN Flash (Thermo Scientific) as reported. 31 BMNs from WT or TRPM2-KO mice were loaded with 2.5 µM Fluo-3 acetoxymethyl (Dojindo laboratories, Kumamoto, Japan) in HEPES-PSS (NaCl 140 mM, KCl 5 mM, CaCl2 1 mM, Glc 10 mM, MgCl2 1 mM, HEPES 10 mM) (Thermo Fisher Scientific) for 30 min at 37°C. After washing twice with HEPES-PSS, 100 µl BMNs (1 × 106/ml) in HEPES-PSS containing 10% FBS were cultured in a 96-well plate. After treatment with 100 nM fMLP at 37°C for corresponding time, fluorescence changes were detected with 490 nm excitation and 526 nm emission wavelengths using a VARIOSKAN Flash.
Bacterial burden determination
Some 16 h after sham or CLP surgery, two separate 3 ml volumes of PBS were injected into the peritoneal space. PLF was harvested by gentle massage of the abdomen for 10 s; 50 µl of the lavage fluid was serially diluted in sterile PBS and 100 µl of the diluent was plated overnight on tryptic soy agar plates at 37°C. Colonies were counted and expressed as log10 CFU/ml of lavage fluid.
Statistical analysis
Data are presented as mean ± SEM. Student’s t-test was used to compare the difference between two groups. One-way analysis of variance was used for multiple comparisons and the Bonferroni post hoc test was used. Survival rate was analyzed by the log-rank test. All data were analyzed using SPSS 17.0 for Windows (SPSS, Chicago, IL). Values of P < 0.05 were considered statistically significant.
Results
Disruption of TRPM2 decreases bacterial clearance in neutrophils
Our previous studies showed that TRPM2 is required for Gram-negative bacterial clearance in macrophages.28,29 To evaluate whether TRPM2 plays a role in bacterial clearance in neutrophils, we performed a bacterial clearance assay. Our data showed that the bacterial burden was greater in TRPM2-KO BMNs than in WT BMNs (Figure 1). This result indicates that TRPM2 plays an important role in bacterial clearance in neutrophils.
TRPM2 deficiency decreases bacterial clearance in neutrophils. 1 × 106 BMNs from WT or TRPM2-KO mice were exposed to E. coli (DH5α) at a BMN/E.coli ratio of 1:20. Cell lysates were plated on Luria–Bertani agar plates and cultured overnight at 37°C to determine bacterial killing capability (n = 4 per group). * P < 0.05, Student’s t-test. Error bars denote the mean ± SEM.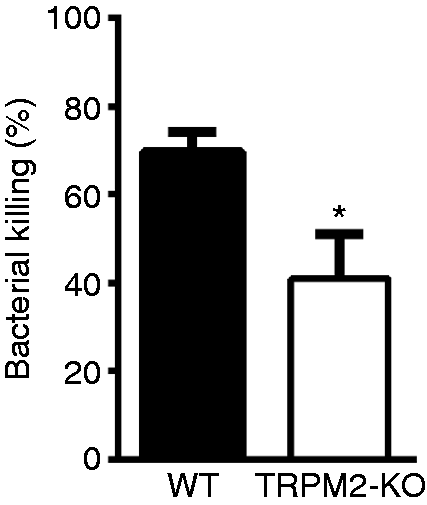
Disruption of TRPM2 decreases elastase release in neutrophils
We then explored the possible mechanism of TRPM2 in bacterial clearance in neutrophils. Elastase is a well-known enzyme released by neutrophils and plays a central role in eliminating invading bacteria.5,6 We investigated the role of TRPM2 in controlling elastase release by using TRPM2-KO neutrophils. We found that the elastase concentration in the supernatant of TRPM2-KO BMNs stimulated with E. coli was lower than that in WT BMNs (Figure 2). These results suggest that TRPM2 plays an important role in bacterial clearance in neutrophils possibly by regulating elastase release.
TRPM2 deficiency decreases elastase release in neutrophils. 1 × 106 BMNs from WT or TRPM2-KO mice were exposed to E. coli (DH5α) at a BMN/E.coli ratio of 1:20 at 37°C for 30 min. Elastase concentration in the supernatant was measured by elastase assay kit (n = 3 per group). *P < 0.05, Student’s t-test. Error bars denote the mean ± SEM.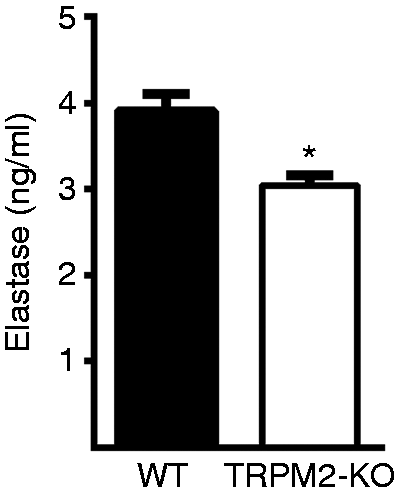
TRPM2-KO mice have decreased elastase release and bacterial clearance after polymicrobial sepsis
We speculated whether TRPM2-KO mice had decreased elastase release and bacterial clearance in the peritoneal cavity after polymicrobial sepsis. As expected, at 16 h after polymicrobial sepsis, elastase concentration increased both in the PLF from WT and TRPM2-KO mice. However, elastase concentration in the PLF from TRPM2-KO mice was significantly lower than that of WT mice (Figure 3a). Bacterial burden in the PLF from TRPM2-KO mice was significantly higher than that of WT mice (Figure 3b). TRPM2-KO mice had decreased survival rate compared with WT mice (Figure 3c). These results further confirmed that TRPM2 plays an important role in bacterial clearance during polymicrobial sepsis, possibly by regulating neutrophil elastase release.
TRPM2–KO decreases elastase release and bacterial clearance after CLP surgery. CLP surgery-induced polymicrobial sepsis was performed in WT and TRPM2-KO mice. (a) Elastase concentration in the PLF from WT and TRPM2-KO mice at 16 h after CLP surgery was measured by elastase assay kit (n = 5 per group). *P < 0.05; ***P < 0.01, One-way analysis of variance. Error bars denote the mean ± SEM. (b) Bacterial burdens in PLF from WT and TRPM2-KO mice at 16 h min after CLP surgery were measured by counting CFUs (n = 6 per group). The CFU of each mouse was represented as one dot. Horizontal bar denotes the means. ***P < 0.001, one-way analysis of variance. (c) Survival was observed after CLP for 7 d (n = 20 per group). *** P < 0.001, Kaplan–Meier log-rank test.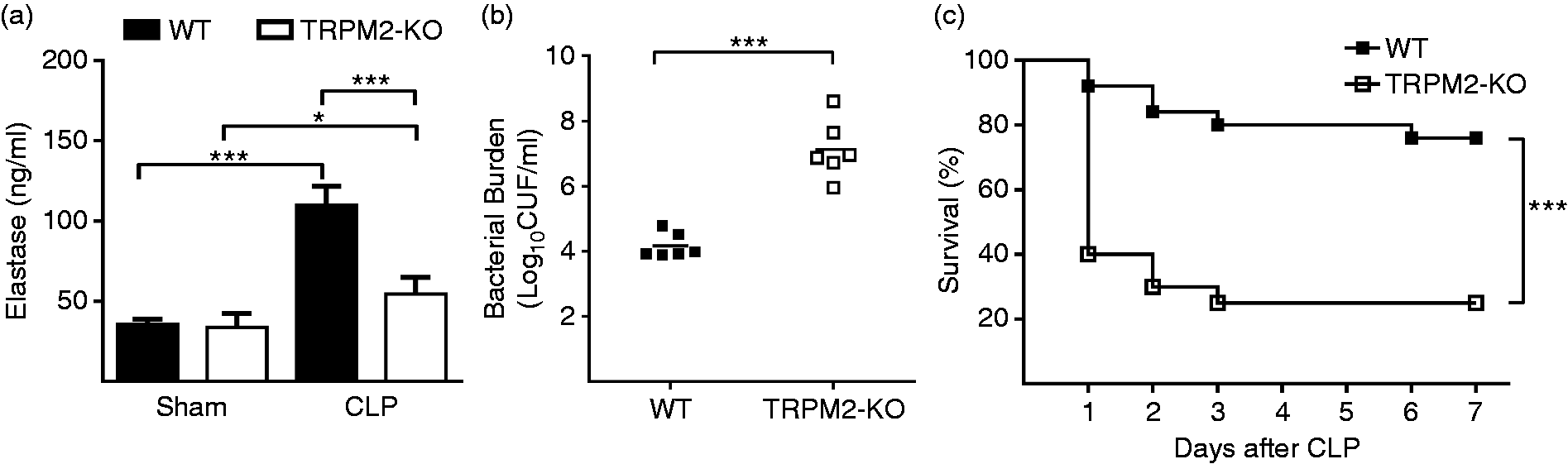
Disruption of TRPM2 attenuates fMLP-induced elastase release in neutrophils
We then examined the role of TRPM2 in regulating elastase release in neutrophils in response to fMLP stimulation. At 10 min after fMLP stimulation, the elastase concentration in the supernatant was significantly lower in TRPM2-KO BDNs than that in WT BDNs (Figure 4). This result indicates that TRPM2 plays an important role in fMLP-induced elastase release in neutrophils.
TRPM2 deficiency decreases fMLP-induced elastase release in neutrophils. 1 × 106 BMNs from WT or TRPM2-KO mice were stimulated with 100 nM fMLP for 10 min at 37°C. Elastase concentration in the supernatant was measured by elastase assay kit (n = 4 per group). *P < 0.05; ***P < 0.001, one-way analysis of variance. Error bars denote the mean ± SEM.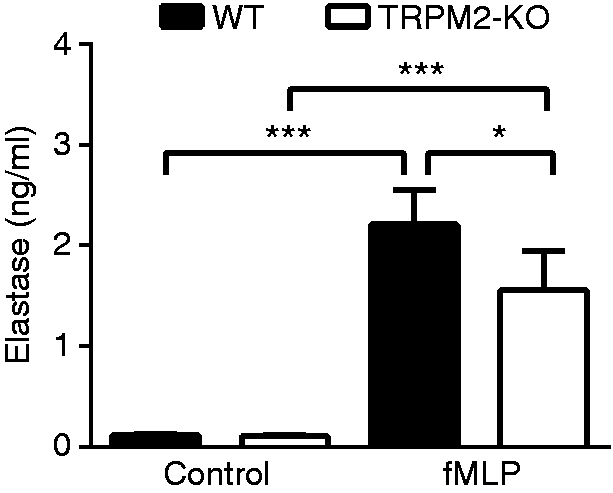
Disruption of TRPM2 attenuates fMLP-induced elastase release in neutrophils partially via decreasing p38 MAPK phosphorylation
Previous studies showed that p38 MAPK plays an important role in elastase release.3,31,33 To confirm whether p38 MAPK contributes to the fMLP-induced elastase release, we pre-treated WT BMNs with p38 MAPK inhibitor at 30 min prior to fMLP stimulation. We found that p38 MAPK inhibitor (SB203580) significantly reduced fMLP-induced elastase release. However, Erk (PD98059) or Jnk (SP600125) inhibitor did not affect fMLP-induced elastase release (Figure 5a and 5b). These results suggest that p38 MAPK plays an important role in fMLP-induced elastase release in neutrophils.
TRPM2 deficiency decreases fMLP-induced elastase release in neutrophils by decreasing p38 MAPKs phosphorylation. (a) Effects of MAPKs inhibitors on extracelluar elastase release in neutrophils in response to fMLP stimulation. WT BMNs were pre-treated with specific inhibitors of the MAPKs (p38 MAPK inhibitor SB203580 (10 µM), Erk inhibitor PD98059 (10 µM), Jnk inhibitor SP600125 (10 µM), or DMSO for 30 min. WT BMNs were then stimulated with 100 nM fMLP for 10 min at 37°C. Elastase concentration in the supernatant was measured by elastase assay kit (n = 4 per group). ***P < 0.001 compared with fMLP treatment, one-way analysis of variance. (b) Effects of the higher concentrations of the Erk and Jnk inhibitors on extracellular elastase release in neutrophils in response to fMLP stimulation. WT BMNs were pre-treated with Erk inhibitor PD98059 (50 and 100 µM) and Jnk inhibitor SP600125 (50 and 100 µM) for 30 min. WT BMNs were then stimulated with 100 nM fMLP for 10 min at 37°C. Elastase concentration in the supernatant was measured by elastase assay kit (n = 3 per group). (c) Effect of TRPM2–KO on p38 MAPK phosphorylation in response to fMLP stimulation in neutrophils. WT or TRPM2–KO BMNs were stimulated with 100 nM fMLP for 10 min at 37°C. Total cell lysates were subjected to immunoblot with anti-phospho-p38 MAPK Ab or anti-p38 MAPK. The p38 MAPK phosphorylation was normalized by total p38 MAPK (n = 3 per group). *P < 0.05; ***P < 0.001, one-way analysis of variance. Error bars denote the mean ± SEM.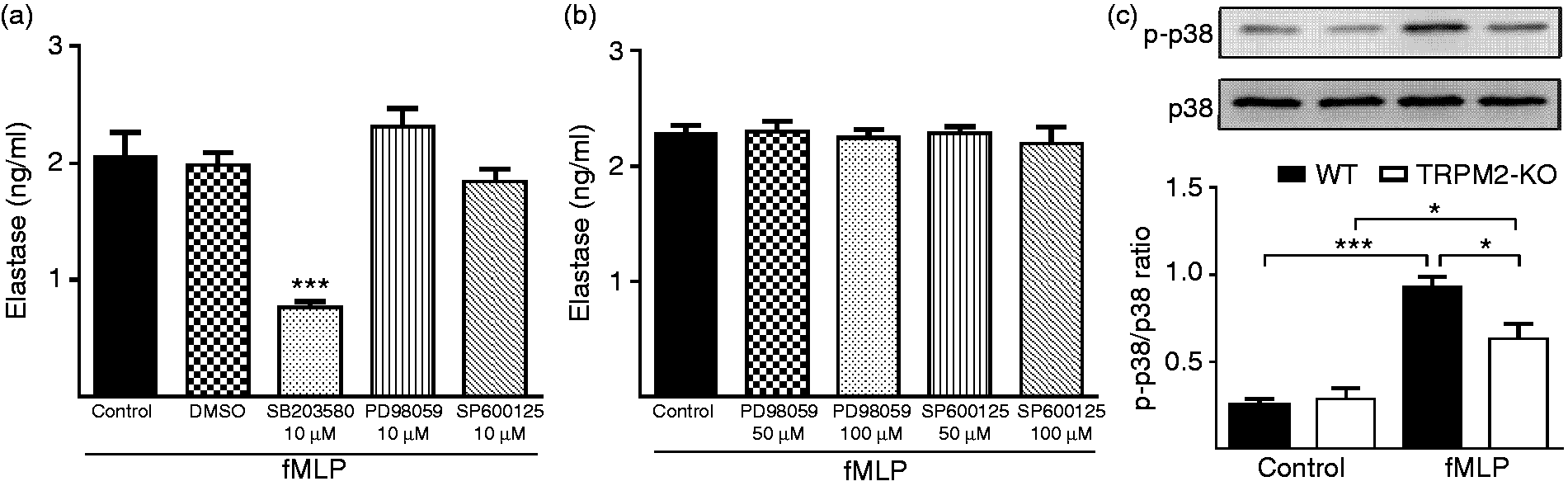
After fMLP stimulation, p38 MAPK phosphorylation was significantly decreased in TRPM2-KO BDNs compared with WT BDNs (Figure 5c). This result indicates that TRPM2 controls fMLP-induced elastase release in neutrophils partially by regulating p38 MAPK phosphorylation.
Disruption of TRPM2 decreases fMLP-induced p38 MAPK phosphorylation in neutrophils possibly via decreasing Ca2+ influx
To investigate whether Ca2+ influx controls fMLP-induced p38 MAPK phosphorylation in neutrophils, extracellular Ca2+ was removed by administrating different concentrations of EGTA before fMLP stimulation. P38 MAPK phosphorylation increased significantly after fMLP stimulation compared with the control. However, EGTA dose-dependently reduced the increased p38 MAPK phosphorylation (Figure 6a). As expected, EGTA also significantly reduced fMLP-induced elastase release (Figure 6b). These results suggest that Ca2+ influx could control fMLP-induced p38 MAPK phosphorylation in neutrophils.
TRPM2 deficiency decreases fMLP-induced p38 MAPK phosphorylation in neutrophils via decreasing Ca2+ influx. (a) Effect of EGTA on p38 MAPK phosphorylation in response to fMLP stimulation in neutrophils. WT BMNs were pre-treated with different concentrations of EGTA for 30 min. WT BMNs were then stimulated with 100 nM fMLP for 10 min at 37°C. Total cell lysates were subjected to immunoblot with anti-phospho-p38 MAPK Ab or anti-p38 MAPK. The p38 MAPK phosphorylation was normalized by total p38 MAPK (n = 3 per group). *P < 0.05; **P < 0.01 compared with control, #P < 0.05 compared with fMLP treatment, one-way analysis of variance. (b) Effect of EGTA on extracellular elastase release from neutrophils in response to fMLP stimulation. After pre-treatment of WT BMNs with or without 1 µM EGTA for 30 min, cells were then stimulated with 100 nM fMLP for 10 min at 37°C. Elastase concentration in the supernatant was measured by elastase assay kit (n = 4 per group). **P < 0.01, Student’s t-test. (c) The changes of intracellular Ca2 + concentration in neutrophils from WT and TRPM2-KO mice after fMLP stimulation. WT or TRPM2-KO BMNs were stimulated with 100 nM fMLP at 37°C at the indicated time points. The changes in fluorescence intensity in Fluo-3AM-loaded BMNs represent changes in intracellular Ca2+ concentration (n = 4 per group). *P < 0.05 compared with WT group, Student’s t-test. Error bars denote the mean ± SEM.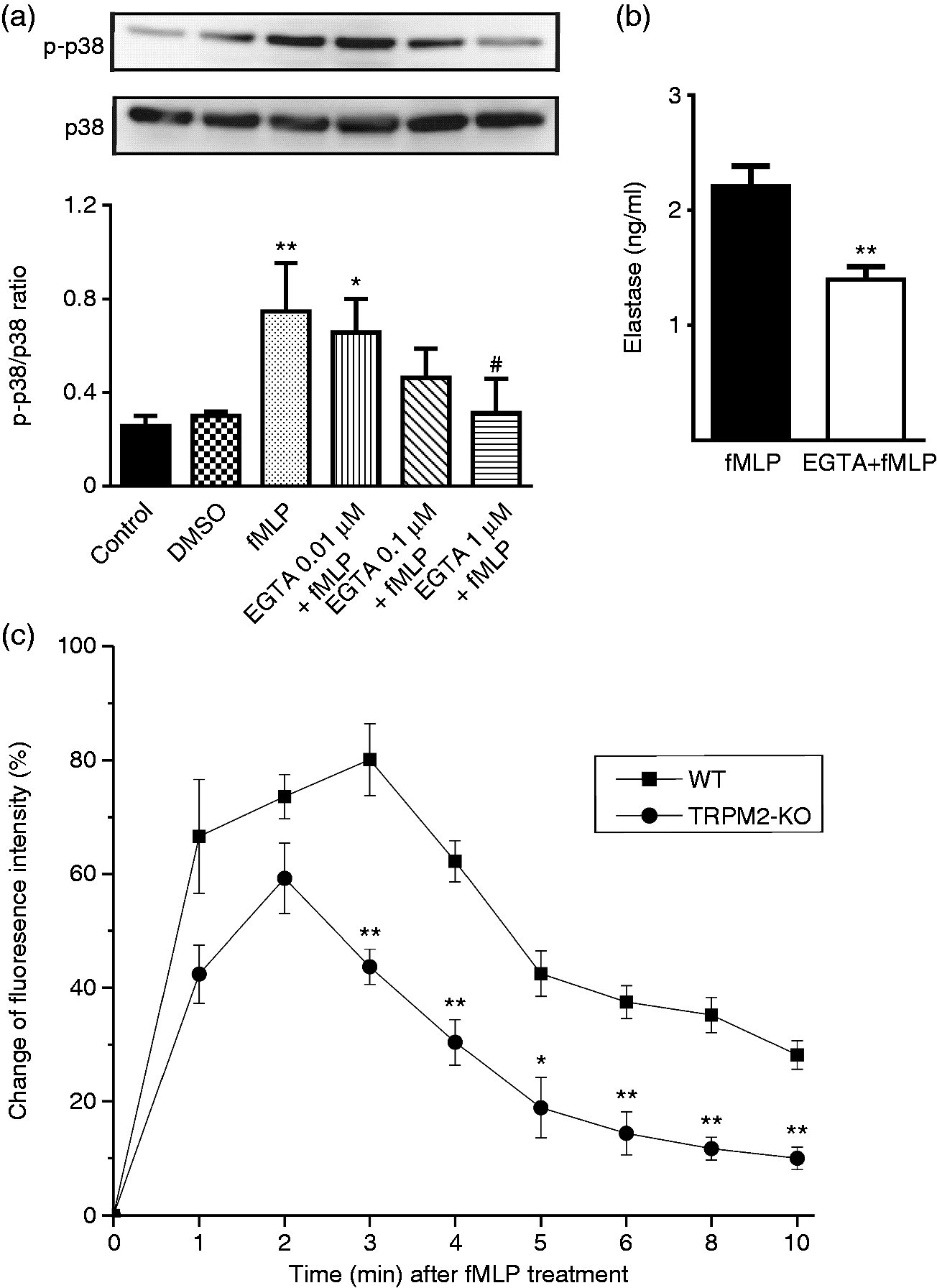
Next, we explored whether genetic disruption of TRPM2 decreases Ca2+ influx in neutrophils after fMLP treatment. We found that the concentration of intracellular Ca2+ decreased significantly in TRPM2-KO BMNs compared with WT BMNs at 3, 4, 5, 6, 8 and 10 min after fMLP treatment (Figure 6c). These data suggest that TRPM2 plays an important role in regulating fMLP-induced p38 MAPK phosphorylation in neutrophils, possibly via regulating Ca2+ influx.
Discussion
The current study demonstrates that TRPM2 is required for bacterial clearance in neutrophils, possibly by regulating elastase release. We found that genetic disruption of TRPM2 decreased bacterial clearance and elastase release in neutrophils treated with E. coli. TRPM2-KO mice also had decreased elastase release, bacterial clearance, and survival rate after CLP-induced polymicrobial sepsis. Our present study also indicates that TRPM2-mediated Ca2+ influx possibly controls fMLP-induced elastase release in neutrophils partially by regulating p38 MAPK phosphorylation.
Neutrophils are recruited to the site of infection and kill invading bacteria which play a critical role in sepsis.34-36 Elastase is a potent serine proteinase released from neutrophil azurophil granules which is critical for host defense against Gram-negative bacteria.5,6 Belaaouaj and colleagues first found that mice deficient in elastase showed increased susceptibility to sepsis and death than WT mice after intraperitoneal injection of Gram-negative bacteria, which suggests that elastase is required by neutrophils to kill intracellular Gram-negative bacteria. 5 In their next research, they explored a mechanism of elastase-mediated clearance of E. coli by degrading the outer membrane protein A on the surface of E. coli. 6 We found that genetic disruption of TRPM2 decreased elastase release from neutrophils exposed to E. coli. In addition, elastase release and bacterial clearance in the PLF from TRPM2-KO mice were also significantly reduced after CLP surgery, which suggests that neutrophil TRPM2 plays an important role in bacterial clearance, possibly by regulating elastase release. Consistent with previous research, Hong and colleagues also observed that shRNA against TRPM2 reduced LPC enhancement of azurophil granule–phagosome fusion and elastase release from neutrophil exposed to E. coli. 31 However, the direct role of neutrophil TRPM2 in elastase release and bacterial clearance has not been explored. Using TRPM2-KO neutrophils and TRPM2-KO mice, the current study demonstrated an important role of TRPM2 in bacterial clearance in neutrophils, possibly by regulating elastase release.
Using fMLP, a strong and specific agonist for neutrophil degranulation, we next attempted to explore the underlying mechanism of TRPM2 in regulating elastase release in neutrophils. Indeed, TRPM2-KO neutrophils showed significantly decreased elastase release in response to fMLP. Previous studies had demonstrated that p38 MAPK is important for neutrophil degranulation.3,31,33 Consistent with previous studies, we also found that only p38 MAPK inhibitor significantly decreased the fMLP-induced elastase release in the WT neutrophils stimulated with fMLP. Genetic disruption of TRPM2 significantly reduced fMLP-induced p38 MAPK phosphorylation in neutrophils. These data indicate that TRPM2 controls elastase release partially by regulating p38 MAPK phosphorylation in neutrophils stimulated with fMLP.
Because Ca2+ influx is important for p38 MAPK phosphorylation in neutrophils,31,33,37 this led us to propose that TRPM2 may control elastase release by regulating p38 MAPK phosphorylation possibly via controlling Ca2+ influx in neutrophils. We observed that fMLP-induced p38 MAPK phosphorylation and elastase release were both reduced by removing extracellular Ca2+. Consistent with previous research,9,23 we also found that disrupting TRPM2 decreased the fMLP-induced increase in the concentration of intracellular Ca2+. Massullo et al. found that Ca2+ influx in mouse neutrophils in response to fMLP was due to activation of TRPM2. 9 In TRPM2-KO mouse neutrophils, fMLP-induced Ca2+ influx and migration were also markedly reduced. 25 Taken together, these data suggest that TRPM2 controls fMLP-induced elastase release by regulating p38 MAPK phosphorylation possibly via controlling Ca2+ influx in neutrophils.
Our present study has some limitations. First, the data from the present study could not elucidate in detail how TRPM2-mediated calcium influx activates p38 MAPK. In human monocytes, calcium influx through TRPM2 is critical for activation of calcium-sensitive tyrosine kinase Pyk2 and downstream Erk signaling. Whether TRPM2-mediated calcium influx activates p38 MAPK directly or indirectly depending on upstream calcium-sensitive tyrosine kinase such as Pyk2 is uncertain. Second, we did not have a neutrophil-specific TRPM2 knockout mouse to study the role of neutrophil TRPM2 in bacterial clearance in sepsis. The genetic deficiency of TRPM2 in multiple tissues still makes it difficult to explore the function of TRPM2 in neutrophils in vivo. Thirdly, we did not investigate whether overexpression of TRPM2 could increase the neutrophil bactericidal activity and whether transfer overexpressed TRPM2 neutrophils into the peritoneal cavity could rescue the septic TRPM2-KO mice. Finally, whether neutrophil TRPM2-mediated elastase, an important enzyme involved in formation of neutrophil nets, plays a role in trapping invading bacteria also needs further investigation.
In summary, our study primarily confirmed that TRPM2 plays an important role in bacterial clearance in neutrophils possibly by regulating elastase release. TRPM2-mediated Ca2+ influx regulates elastase release partially via p38 MAPK phosphorylation in neutrophils. Our data add an additional mechanism regarding the protective role of TRPM2 in polymicrobial sepsis. The underlying mechanism of TRPM2 in regulating neutrophil bacterial clearance is largely unexplored and requires further investigation.
Footnotes
Acknowledgments
We thank Prof. Y. Mori (Graduate School of Engineering, Kyoto University, Katsura Campus, Kyoto, Japan) for providing the TRPM2-knockout mice.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the Zhejiang Provincial Natural Science Foundation of China under grant NO.LY15H150007; and the National Natural Science Foundation of China under grant NO.81501702.