
Abstract
Mutations in the nucleotide binding domain of the PRR, NOD2, are associated with the autoinflammatory diseases Blau syndrome and early-onset sarcoidosis. Current theories suggest that constitutive activation of the NOD2 pathway may be responsible for pathogenesis of these diseases. Here, we report the phenotype of a kindred with Blau syndrome caused by a novel NOD2 mutation (p.E383D). Signaling protein and cytokine expression were examined, and the results of these experiments challenge current theories of constitutive NOD2 activation in the pathophysiology of Blau syndrome.
Introduction
Blau syndrome (BS) and early-onset sarcoidosis (EOS) are familial and sporadic forms, respectively, of an autosomal dominant disease classically manifesting with arthritis, uveitis and rash. These diseases are thought to arise as consequence of gain-of-function mutations in NOD2. NOD2 activation is thought to exert its impact via two major signaling pathways, namely the MAP kinase pathway and the NF-κB pathway, which regulate the expression of chemokines and inflammatory cytokines. In vitro studies in cell lines transfected with nucleotide binding domain (NBD) mutations previously reported in patients with BS and EOS have found increased activity of NF-κB in the absence of stimulation,1–4 suggesting constitutive NOD2 activation may underlie the pathogenesis of these diseases. However, studies of cytokine production in patients with BS have not provided support for this theory.5–7 Here, we report a novel mutation in NOD2 in a kindred previously described by Ting et al., 8 who were diagnosed on the basis of clinical and histopathological findings. We update the clinical phenotype of this family and examine the functional consequence of the novel mutation utilizing in vivo and ex vivo methods.
Case reports
P1, a 45-yr old female, was diagnosed with juvenile idiopathic arthritis (JIA) at 2 yr of age. She was the first patient with BS documented to have renal granulomas, interstitial nephritis, corticomedullary fibrosis and acute kidney injury. As a 43-yr old, P1 developed abnormal liver function tests with a cholestatic pattern, despite previous cholecystectomy. Liver biopsy showed cirrhosis and hepatic granulomas. She had not developed decompensated liver disease. Her medications included prednisolone, ursodeoxycholic acid, rabeprazole, salbutamol and fluticasone/salmeterol inhaler.
The daughter of P1 and index patient in the report by Ting et al., 8 P2, is a 24-yr old who was diagnosed with BS at 3 yr of age after developing arthritis and rash. Biopsy of the rash showed granulomas in the superficial and papillary dermis. At review, her medications included methotrexate, prednisolone, folic acid, inhaled fluticasone/salmeterol, salbutamol, cetirizine and prochlorperazine.
P3, the son of P1, is an 18-yr old male who was diagnosed with BS in the first yr of life following biopsy of a rash in the context of a positive family history. There was associated arthritis affecting the shoulders, hands, wrists, knees and ankles, with significant early morning stiffness. He had previously been treated with corticosteroid injections to the knees, wrists and ankles, and was commenced on methotrexate at 9 years of age. His past medical history includes asthma, supraventricular tachycardia, anxiety and childhood attention hyperactivity disorder. He has not had a myocardial biopsy. At review, his medications included methotrexate, folic acid and salbutamol.
None of the patients displayed evidence of BS activity at clinical review. Patients provided written informed consent for the publication of the preceding case reports.
Materials and methods
NOD2 genotyping
Written informed consent was obtained from patients and controls for analyses approved by the Western Sydney Local Health District Human Research Ethics Committee [HREC2002/9/3.6(1425)]. DNA was extracted from whole blood using the QIAsymphony DSP DNA Mini Kit (Qiagen, Hilden, Germany) as per the manufacturer’s instructions. Exon 4 of NOD2 (GenBank accession no. NC_000016) was amplified by PCR and analyzed by automated sequencing. PCR was carried out with HotStarTaq DNA polymerase kit (Qiagen, Crawley, UK). Owing to the large size, NOD2 exon 4 was amplified in three reactions with the use of the following primers: fragment 1 forward primer 5′-CAGCTTGTGAATGGAGGAGC-3′ and reverse primer 5′- GCTGGTCACCACCTTGC-3′; fragment 2 forward primer 5′- TTTCTCTTTGTCTTCCCATTC-3′ and reverse primer 5′- GGCAAAGAAGCACTGGAAAG-3′ and fragment 3 forward primer 5′- GGGCAGACTGGCTCTGTG-3′ and reverse primer 5′- GCCCAGACTTCTAGAGCCC-3′.
The PCR cycling conditions were as follows: initial denaturation at 95℃ for 15 min, then 35 cycles consisting of denaturation at 94℃ for 45 s, annealing at 60℃ for 45 s and extension at 72℃ for 1 min, followed by a single cycle of 72℃ for 10 min. PCR products were purified with a QIAquick PCR purification kit (Qiagen) according to the manufacturer’s protocol and sequenced with the Big Dye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) following the procedures recommended by the manufacturer. The sequence of NOD2 was analyzed on the ABI 3130xl Genetic Analyser using Sequencing Analysis Software version 5.4.
Western blot analysis
PBMCs from P1–P3, three healthy controls (C1–C3) and two treatment controls on combination methotrexate and prednisolone therapy (T1 and T2) were isolated using Percoll gradient centrifugation and re-suspended in RPMI + 10% FBS. For analysis of NF-κB and MAPK activation, 5 × 106 cells were lysed in 250 µl sample buffer and subjected to Western blot analysis. Briefly, 12 μl lysate/lane were separated on a SDS-PAGE gel (8–12% gradient) and transferred to PVDF membranes for Ab detection of markers for NF-κB and MAPK activation, respectively. Abs against P-p65 (#3033), IκBα (#9242), P-IκBα (#9246), P-ERK (#9101) and P-p38 (#4511) were purchased from Cell Signaling Technology (Danvers, MA, USA).
Cytokine analysis
For cytokine analysis, 2 × 105 cells from individuals P1–P3 and C1–C3 were stimulated with either the NOD2 ligand L18-muramyl dipeptide (MDP; 200 ng/ml; Invivogen, San Diego, CA, USA) or the TLR4 ligand ultrapure LPS (25 ng/ml; Invivogen) for 16 h. Supernatants were harvested, cleared of cellular debris and cytokine concentrations measured by multiplex analysis (BioRad, Hercules, CA, USA). Samples were measured in triplicate and the mean was calculated for each sample. Differences in cytokine concentrations between subjects and controls were compared using ranked t-tests. 9 To determine the contribution of the effects of immunosuppression on cytokine production, the analysis was subsequently repeated with the inclusion of T1 and T2.
Results
Sequencing of exon 4 of NOD2 revealed a novel mutation (c.1149G > T; Figure 1) leading to substitution of the highly conserved glutamic acid residue with aspartic acid at position 383 (p.E383D) in all affected individuals. No mutations in NOD2 were found in the unaffected brother of P1.
Partial sequence of NOD2 showing (A) DNA from the negative control and (B) DNA from patient 1 diagnosed with BS caused by c.1149G > T substitution resulting in the E383D mutation.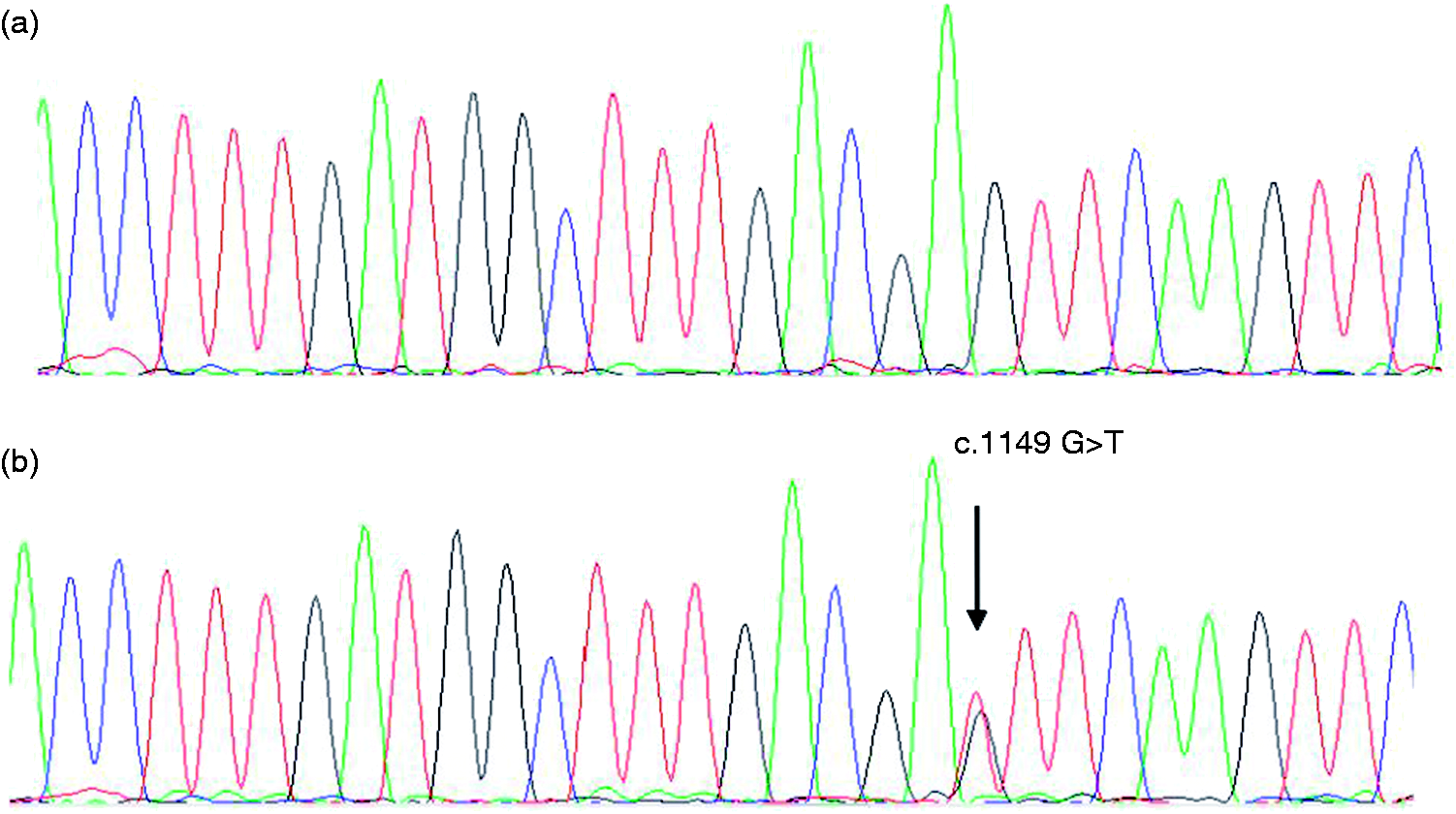
Western blot analyses of unstimulated PBMCs from our kindred, three healthy controls and two treatment controls were compared (Figure 2). Decreased activation of NF-κB was observed in affected individuals and treatment controls in comparison with healthy controls. In contrast, levels of p38 phosphorylation were similar among all the samples. Densitometry measurements for Western blot analyses were normalized against GAPDH and were consistent with the abovementioned findings (data not shown).
Patients with NOD2 E383D have decreased NF-κB activation and increased levels of P-ERK. Equal numbers of freshly isolated PBMCs from healthy donors (C1–C3), patients with BS (P1–P3) and treatment controls (T1 and T2) were lysed and loaded onto SDS-PAGE. This was followed by Western blot for activation markers of NF-κB and MAP kinase. Levels of P-p65 and P-IκBα are decreased in patient samples and treatment controls vs. healthy controls. Levels of P-p38 were similar between patients and controls. Total GAPDH levels act as a loading control. The figure represents a single Western blot experiment.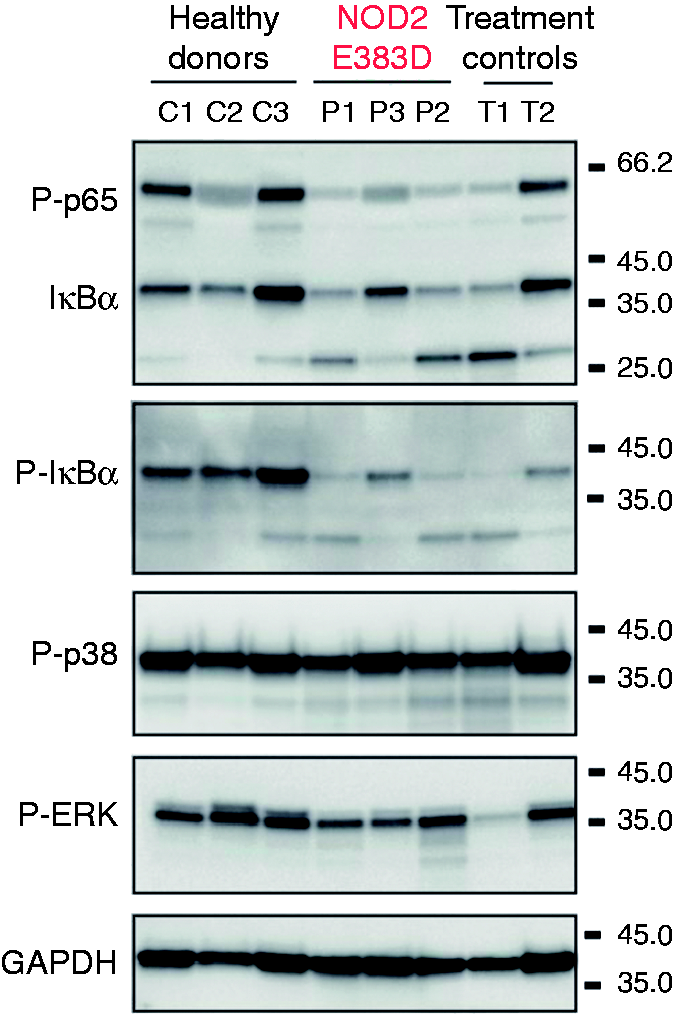
Consistent with low levels of NF-κB activity in patient cells, baseline concentrations of inflammatory cytokines, IL-1β, IL-6, TNF-α and IFN-γ were significantly reduced when compared with cells from healthy donors (Figure 3). Interestingly, stimulation of PBMCs with the NOD2 ligand, MDP, resulted in increased cytokine production from healthy control cells; however, this response was severely attenuated in BS patient cells. In contrast, LPS (TLR4 ligand) stimulation resulted in robust cytokine expression in all samples, suggesting that the hypo-responsiveness in patients with BS is specific for NOD2 and not due to generalized cellular unresponsiveness. Quantitative PCR of RNA encoding TNF and IL-1β in unstimulated PBMCs was consistent with this, with decreased expression in patients with BS compared with controls (data not shown).
NOD2 E383D results in hypo-responsiveness to MDP stimulation. Freshly isolated PBMCs from three healthy donors (C1–C3, white symbols) and three patients with BS (P1–P3, black symbols) were either left untreated, treated with L-18 MDP or with LPS and cytokines were measured in triplicate after 16 h. Patients with BS displayed a normal response to LPS, but demonstrated blunt responses to L-18 MDP stimulation. OOR: out of range (>5000 pg IL-6/ml); wt: healthy controls; ‘control’: untreated. *Statistically significant difference at the 0.02 significance level.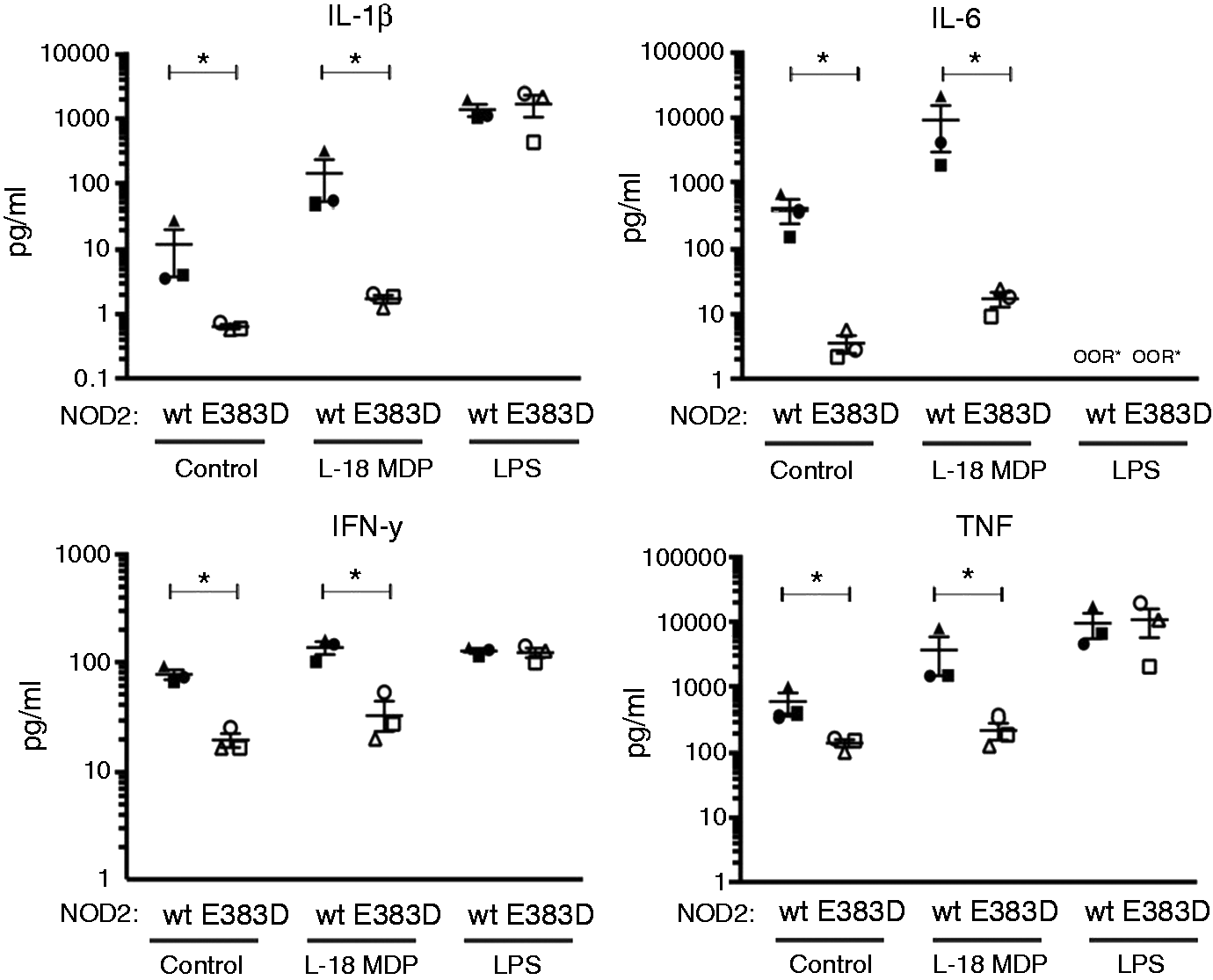
To take into account differences in baseline cytokine production, the absolute difference of cytokine concentrations for L18-MDP stimulated cells to unstimulated cells was compared between subjects and controls using ranked t-tests. Across all of the assayed cytokines, L18–MDP stimulation resulted in significantly greater increases in cytokine production in the healthy control group compared with patients with BS (P = 0.02 for all comparisons).
In order to determine whether hypo-responsiveness to MDP was due to the functional effects of NOD2 mutation or chronic immunosuppression, cytokine stimulation experiments were repeated with treatment controls who were on prednisolone and methotrexate for sarcoidosis. For technical reasons, only IL-6 levels were reliably quantified. IL-6 production appeared to be severely depressed in patients with BS vs. other groups (Figure 4).
NOD2 E383D hypo-responsiveness to MDP stimulation is not due exclusively to immunosuppression. Cytokine stimulation was repeated with the addition of treatment controls on prednisolone and methotrexate for sarcoid. IL-6 concentrations were measured and showed robust responses in these individuals whilst being depressed in patients with BS. Treatment controls responded robustly to LPS stimulation.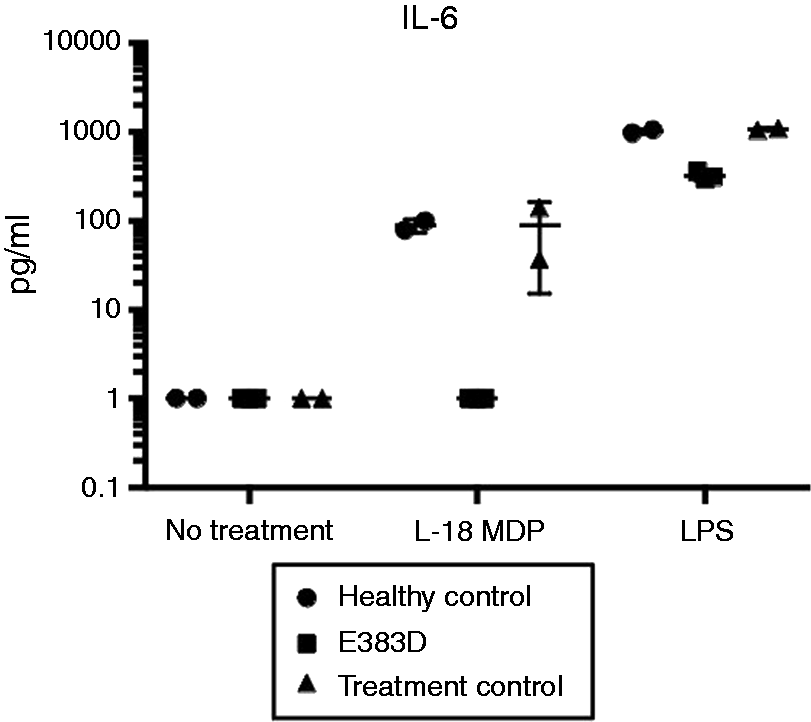
Discussion
Several mutations in NOD2 are known to be associated with BS and EOS. These mutations have been localized to the NBD, which is believed to be responsible for NOD2 oligomerization and subsequent recruitment of receptor interacting protein kinase 2. The novel mutation identified at position 383 is located at a highly conserved region of the NBD, thought to participate in the hydrolysis of ATP and hence NOD2 deactivation. 10 Mutations at this site have also been previously described in patients with BS/EOS.11–15
In vitro studies in cell lines transfected with NBD mutations identified in patients with BS and EOS have reported increased activity of NF-κB in the absence of stimulation,1–4 suggesting constitutive NOD2 activation may underlie the pathogenesis of these diseases. Our results, however, do not support this theory. We found low basal NF-κB expression and, accordingly, low cytokine levels in patients with NOD2 mutations vs. healthy controls. Even with concurrent immunosuppression, non-specific stimulation of NOD2 mutant PBMCs with LPS resulted in robust increases in cytokine production, suggesting that depressed MDP stimulated responses were not due exclusively to concurrent use of immunosuppression. In addition, treatment controls on similar immunosuppressants did not display such depressed IL-6 responses to MDP, further supporting this finding.
Negative feedback pathways that attenuate downstream signaling and cytokine production may account for clinically inactive disease and the results obtained in our BS cohort. For example, NOD2 activation is hypothesized to result in its dissociation from HSP90 and subsequent proteasomal degradation. SOCS3, which is induced on exposure to MDP, may also play a role in the ubiquitination of NOD2 and contribute to its proteasomal degradation. 16 Further downstream, the NF-κB pathway is subject to a myriad of positive and negative regulatory pathways (see Boyle et al. 17 for a review), which may account for decreased protein and cytokine expression found in our study. These regulatory mechanisms might also provide a unifying explanation for conflicting results of studies on BS/EOS pathophysiology. It is quite plausible that the state of regulatory pathways in NBD mutant cell lines does not reflect those seen in patients with BS/EOS from which ex vivo studies have been conducted. Comprehensive studies that assess the regulatory pathways in addition to protein and cytokine expression would assist in clarifying this issue.
In summary, we have identified a novel NOD2 mutation that accounts for the clinical features of a kindred with BS. Functional studies including protein and cytokine expression profiles have also been obtained for this mutation. Although the condition is thought to result from constitutive activation of NOD2 and increased NF-κB pathway signaling, our results argue against this. This raises interesting questions for the field of BS/EOS pathophysiology, where gain of function NOD2 is the currently accepted dogma. 18 Firstly, the mechanism leading to apparent NOD2 hypofunction is unclear. Mice possessing the R314Q mutation (equivalent to the BS associated mutation R334Q in humans), have a hypo-functioning, truncated NOD2 molecule, which is thought to result from a post-translational modification. 7 Whether this occurs in humans is unclear, and warrants further investigation. Any model would also need to explain the dominant negative effects of mutant NOD2 on unmutated, activated NOD2 dimers or combination mutant/non-mutant heterodimers. Secondly, the clinical manifestations of BS/EOS would not be explained by hypo-functioning NOD2 and requires further examination. Future studies should consider a longitudinal, global examination of the NOD2 pathway and its regulatory components to more clearly elucidate the pathophysiology of BS and EOS at various states of clinical activity. Such an understanding may provide useful biomarkers of disease activity and would pave the way for treatments that have precise but profound effects for sufferers of these diseases.
Footnotes
Acknowledgements
The authors would like to thank Dr. Helen Lachmann of the National Amyloidosis Centre, UK, for facilitating NOD2 sequencing and Aleksandra Bankovacki for assistance with Western Blot and cytokine stimulation experiments The late Dr. Brien Walder made a clinical diagnosis of BS in patient P2 a short time after it was first reported and recognized that the diagnosis of JIA in P1 should be revised.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by an National Health and Medical Research Council (Project Grant #1046986); a Future Fellowship from the Australian Research Council to UN (FT 130100166); and was made possible through Victorian State Government Operational Infrastructure Support and Australian Government IRIISS (#9000220).