
Abstract
This study investigated the pathogenesis of periodontitis and the role of nucleotide-binding oligomerization domain-like receptor protein 10 (NLRP10). The human oral epithelial cell line HOK-16B was infected with two periodontal pathogens, Tannerella forsythia and Fusobacterium nucleatum, at various MOIs. RT-PCR and immunoblotting demonstrated that infection increased mRNA and protein expression of NLRP10, respectively. The siRNA-mediated NLRP10 knockdown significantly reduced IL-1α expression and secretion. Both bacteria induced phosphorylation of ERK, JNK and p38 MAP kinases in HOK-16B cells. NLRP10 knockdown impaired ERK phosphorylation only. ERK inhibition significantly decreased the expression of T. forsythia- and F. nucleatum-induced IL-1α. Our data suggest that NLRP10 is involved in activating the ERK signalling pathway in HOK-16B cells infected with T. forsythia and F. nucleatum. This pathway likely augments the pro-inflammatory cytokine IL-1α levels, which may play a critical role in periodontitis.
Introduction
Innate immunity is the front line of host defence against pathogens. The host innate immune system evolved to cope with pathogens that adopted escape-avoidance strategies for survival. NLR proteins are PRRs that serve as intracellular sensors for microbial- and host-derived components.1,2 Several NLR proteins are components of the inflammasome that is linked to caspase-1 activation, which results in the maturation and secretion of IL-1β and IL-18.
NLRP10 is the smallest human NLR protein, and it differs from the other NLR proteins because it lacks the leucine-rich repeat domain, which participates in ligand sensing or binding. NLRP10 is expressed in various human and murine cells.3,4 The functional role of NLRP10 was not as intensively studied as the other NLR proteins, including NOD1, NOD2, NLRP1 and NLRP3. 5 However, anti- and pro-inflammatory functions of NLRP10 were reported. NLRP10 inhibited ASC-mediated NF-κB activation, caspase-1-dependent IL-1β release and cell death in HEK293 cells transfected with plasmid DNA containing an NF-κB enhancer and luciferase gene.3,6 Reduced IL-1β secretion from macrophages of NLRP10-transgenic mice was observed after stimulation with Salmonella Typhimurium or the TLR7 R837 ligand. 7 NLRP10-transgenic mice were resistant to endotoxic shock. 7 Amyloid-β-induced cathepsin expression in rat glial cells led to NLRP10 degradation and subsequent NLRP3 inflammasome formation. 8 In contrast to these anti-inflammatory features, NLRP10-deficient mice failed to induce specific Th1 and Th17 responses against Candida albicans infection. 9 NLRP10 knockdown in HeLa cells infected with Shigella flexneri reduced pro-inflammatory cytokine expression, including IL-6 and IL-8. 10 NLRP10 co-localized with NOD1 at the site of bacterial entry in HeLa cells and interacted with ectopically expressed NOD1 in HEK293T cells. 10
Periodontitis is a chronic oral inflammatory disease that is the result of excessive host immune and inflammatory responses to bacteria in the subgingival sulcus. Periodontitis may cause alveolar bone and tooth loss. 11 The presence of major periodontal pathogens, including Treponema denticola, Tannerella forsythia and Porphyromonas gingivalis, increases the risk of periodontitis. 12 These three organisms constitute the so-called ‘red complex’. Fusobacterium nucleatum also plays a critical role in the development of pathogenic subgingival biofilm via the co-aggregation of periodontal pathogens with early-colonized bacteria. Periodontitis is also associated with systemic diseases, including atherosclerosis, rheumatoid arthritis and preterm birth.12–14 Recent studies demonstrated that NLR proteins play an important role in the inflammation induced by periodontal pathogens. NLRP3 inflammasome activation by P. gingivalis, T. denticola and Aggregatibacter actinomycetemcomitans induced IL-1β secretion in monocytes and macrophages.15–18 Porphyromonas gingivalis in subgingival biofilm down-regulated NLRP3 expression and IL-1β secretion, which prolonged the survival and persistence of biofilm species during the host immune response. 19 Fusobacterium nucleatum induced IL-1β secretion via NLRP3 activation in gingival epithelial cells. 20 A. actinomycetemcomitans increased NLRP3 gene expression and reduced NLRP6 expression, but had no effect on NLRP1 or NLRP2 expression. 21 Outer membrane vesicles of A. actinomycetemcomitans were internalized and activated NOD1- and NOD2-dependent NF-κB signalling pathways in HEK293T cells. 22 NOD1 signalling induced alveolar bone loss in a mouse model of periodontitis. 23 T. forsythia induced the release of endogenous danger molecules from macrophages, possibly via an inflammasome-dependent manner. 24 However, the expression and role of NLRP10 were not investigated in periodontal tissues.
The present study examined the innate immune responses of NLRP10 in the HOK-16B oral epithelial cell line after infection with T. forsythia and F. nucleatum. Both of these organisms increased NLRP10 expression, which was associated with ERK activation and a subsequent upregulated production of the pro-inflammatory cytokine IL-1α. These results suggest that NLRP10 exhibits a novel function against periodontal pathogens in the pathogenesis of periodontitis.
Materials and methods
Reagents and Abs
The Easy-BLUE total extraction kit was purchased from iNtRON Biotechnology (Sungnam, Korea). Penicillin–streptomycin, carboxylfluorescein diacetate succinimidyl ester (CFSE), NLRP10 Stealth siRNA, Stealth siRNA negative control duplex and Lipofectamin RNAiMAX were purchased from Invitrogen (Carlsbad, CA, USA). Recombinant IL-1α, PD 98059 (an ERK inhibitor) and Ac-YVAD-CHO (a caspase-1 inhibitor) were purchased from Biolegend (San Diego, CA, USA). An LDH cytotoxicity assay kit was purchased from Biovision (Palo Alto, CA, USA). An anti-NLRP10 Ab (cat. no. MABC293) was purchased from Merck Millipore (Billerica, MA, USA). 3,3′,5,5′-Tetramethylbenzidine (TMB), gentamicin and metronidazole were purchased from Sigma (St. Louis, MO, USA). Rabbit polyclonal anti-caspase-1 (cat. no. 2225), anti-IκB-α (cat. no. 9242), anti-phospho-SAPK/JNK (Thr183/Tyr185) (cat. no. 9251), anti-phospho-p44/42 (Thr202/Tyr204) (cat. no. 9101) and anti-phospho-p38 (Thr180/Tyr182) (cat. no. 9211) Abs were purchased from Cell Signaling Technology (Beverly, MA, USA). Goat polyclonal anti-IL-1β (cat. no. sc-1250), rabbit polyclonal anti-IL-1α (cat. no. sc-7929) and HRP-conjugated anti-rat IgG (cat. no. sc-2065) Abs were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Mouse monoclonal anti-β-actin (cat. no. 612656), HRP-conjugated anti-rabbit IgG (cat. no. HAF008), anti-goat IgG (cat. no. HAF109) and anti-mouse IgG (Cat. no. HAF007) Abs were purchased from R&D Systems (Minneapolis, MN, USA).
Bacterial strains and growth conditions
T. forsythia (ATCC 43037) was grown in new oral spirochete medium (ATCC medium 1494) supplemented with 0.01 µg/ml N-acetylmuramic acid. F. nucleatum (ATCC 25586) was grown in a brain heart infusion broth (BD Bioscience, San Jose, CA, USA) under anaerobic conditions (10% H2, 10% CO2, and 80% N2). Streptococcus oralis was aerobically grown in Tryptic Soy Broth (BD Bioscience). The number of bacteria was calculated using pre-made standard curve (OD value – CFU) and diluted in antibiotic-free media.
Cell culture and treatment
HOK-16B cells are an immortalized human epithelial cell line that originated from Dr. Park at the University of California, Los Angeles. 25 The cells were grown in keratinocyte growth medium containing a supplementary growth factor bullet kit (Lonza, Walkersville, MD, USA). HOK-16B cells (5 × 105/ml) were cultured in six-well plates (Becton Dickinson Labware, Franklin Lakes, NJ, USA) for 24 h and washed with antibiotic-free medium prior to bacterial infection. Bacteria were added to the HOK-16B cells at MOIs of 10, 100, 500 and 1000 for various infection times. The cells were harvested for gene and protein expression analyses. Supernatants were obtained for ELISA and immunoblotting analyses.
Real-time RT-PCR
RNA was extracted from HOK-16B cells using the easy-BLUE™ total RNA extraction kit. The cDNA was synthesized from 1 µg of RNA using an M-MLV reverse transcriptase kit (Promega, Madison, WI, USA). Real-time RT-PCR for NLRP10, IL-1α, IL-8, COX-2 and GAPDH was performed using reagents from the Power SYBR® Green Master mix (Applied Biosystem, Warrington, UK). PCR was repeated for 40 thermal cycles: denaturation at 95℃ for 15 s and annealing/extension at 60℃ for 1 min. GAPDH expression was used to normalize gene expression. The following primer sequences were used: 5′-GTT GGA GGG CCT GAT TCC GGT G-3′ and 5′-GCA GCG CAC ATG CTC TCG GTA T-3′ for NLRP10; 5′-GTT TAA GCC AAT CCA TCA CTG ATG-3′ and 5′-GAC CTA GGC TTG ATG ATT TCT TCC T-3′ for IL-1α; 5′-CTG TGT GAA GGT GCA GTT TTG-3′ and 5′- AAC TTC TCC ACA ACC CTC TGC-3′ for IL-8; 5′-CAA ATT GCT GGC AGG GTT GC-3′ and 5′-TCA CCA TAG AGT GCT TCC AAC TC-3′ for COX-2; 5′-GTC GCC AGC CGA GCC-3′ and 5′-TGA AGG GGT CAT TGA TGG CA-3′ for GAPDH.
Immunoblotting
HOK-16B cells infected with T. forsythia, F. nucleatum and S. oralis were harvested, washed with DPBS, and lysed in 30 µl of RIPA buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 50 mM NaF, 1 mM EDTA, 5 µM Na3VO4 and 1 mM PMSF). Lysates were obtained after centrifugation for 45 min at 16,000 g and 4℃. T. forsythia or F. nucleatum pellets (2.5 × 108 bacteria) were pulsed with sonicator for 30 s five times in order to break bacterial cell walls. The pellets were then lysed in 20 µl of RIPA buffer. Supernatants and bacteria culture medium were precipitated with a 6.1 N trichloroacetic acid solution (Sigma-Aldrich). Precipitates were separated from supernatants using further centrifugation for 10 min at 16,000 g and 4℃. Precipitates were neutralized with 0.1 N NaOH. Samples were prepared via the addition of a 5× SDS sample buffer. Cell lysates and TCA-precipitated proteins were separated using SDS-PAGE (12% polyacrylamide gel) and transferred onto PVDF membranes. Membranes were blocked and incubated with primary Abs targeting specific proteins for 1–2 d. Membranes were washed with PBS-1% Tween 20 (AMRESCO, Solon, OH, USA) and incubated with a HRP-conjugated secondary Ab. Proteins were detected using the SUPEX detection reagent (Dyne-Bio, SeongNam, Korea) in a chemiluminescence imaging chamber.
Indirect ELISA
IL-1α levels were measured in the culture supernatants of the infected HOK-16B cells. Culture supernatants and serially diluted recombinant IL-1α for the standard were added to 96-well plates and incubated overnight. The wells were washed and blocked with 1% BSA in PBS for 1 h. The wells were incubated with an anti-IL-1α Ab for 2 h. The wells were washed and incubated with a HRP-conjugated anti-rabbit IgG Ab for 2 h. The wells were incubated with TMB in phosphate-citrate buffer for 20 min. A stop solution was added to the wells, and the samples were analysed for absorbance at 450 nm.
RNA interference assay
Pre-designed Stealth siRNAs for NLRP10 (NM_176821) was purchased from Invitrogen (Carlsbad, CA, USA). The following sequences for the duplexes were used: 5′-GGA AUU CUC CCU GUA UGA AGC UAA A-3′ and anti-sense: 5′-UUU AGC UUC AUA CAG GGA GAA UUC C-3′ for NLRP10. Specific and control siRNAs were adjusted to a concentration of 30 nM and mixed with the transfection reagent, Lipofectamin RNAiMAX. This mixture was diluted in 250 µl of gentamicin-free HOK-16B cell culture medium for 24 h in a 37℃ incubator with 5% CO2. Cells were washed with antibiotic-free medium and infected with T. forsythia and F. nucleatum for various times. Knockdown of NLRP10 was confirmed using real-time RT-PCR and immunoblotting.
Inhibitor assay
HOK-16B cells were pre-incubated with an ERK inhibitor (PD98059) and caspase-1 inhibitor (Z-YVAD-FMK) diluted in 900 µl of antibiotic-free medium for 30 min. The cells were infected with T. forsythia and F. nucleatum at a MOI 500 in 100 µl of medium for 6 h.
LDH assay
HOK-16B cells (5 × 104 cells/well) in 96-well plates were infected with T. forsythia or F. nucleatum for 6 h. LDH release into culture supernatants was measured using an LDH cytotoxicity assay kit (BioVision, Palo Alto, CA, USA) according to the manufacturer's instructions. Cells were lysed with 1% Triton X-100 to determine maximal LDH release (100%).
Invasion assay
HOK-16B cells (1 × 105cells/well in 24 well plates) were transfected with NLRP10 siRNA or control siRNA for 24 h and infected with CFSE-labelled T. forsythia and F. nucleatum for 6 h. Cells were washed with DPBS three times and detached using Trypsin-EDTA. The fluorescence of the cells was analysed using flow cytometry (FACSCalibur; BD Bioscience). Intracellular bacteria were detected after quenching extracellular fluorescence with 400 µg/ml trypan blue.
Statistics
Samples were compared using standard two-tailed Student’s t-test. The P-values are indicated in the figure legends.
Results
T. forsythia and F. nucleatum enhanced NLRP10 expression
We investigated the role of periodontal pathogens on NLRP10 expression in HOK-16B cells to elucidate the role of NLRP10 in inflammation. We analysed the expression of NLRP10 after bacterial infection at various time points. NLRP10 expression increased significantly from 3 to 12 h (data not shown). We chose the time point of 6 h after infection to analyse protein and mRNA expression. We found that T. forsythia and F. nucleatum up-regulated NLRP10 mRNA expression in an MOI-dependent manner using real time RT-PCR (Figure 1a). NLRP10 protein level similarly increased in the presence of these bacteria using immunoblotting (Figure 1b). Notably, increased NLRP10 levels were detected in culture supernatants. We also included the oral commensal bacteria S. oralis, which did not induce NLRP10 expression (Figure 1c). These results demonstrated a pathogen-specific NLRP10 up-regulation in HOK-16B cells. To confirm the specificity of the anti-NLRP10 Ab used in this study, immunoblot analysis was performed on bacterial lysates and culture medium of HOK-16B cells. The NLRP10 Ab did not cross-react with bacterial proteins or with components from the culture medium of HOK-16B cells (Figure S1).
NLRP10 mRNA and protein were increased in HOK-16B cells infected with T. forsythia and F. nucleatum. HOK-16B cells (5 × 105) were infected with T. forsythia, F. nucleatum or S. oralis for 6 h at various MOIs. (a and upper panel of c) The expression of NLRP10 mRNA was analysed using real-time RT-PCR (right panel of a). The data are shown as the means ± SD of three experiments performed in triplicate. RT-PCR products were observed on agarose gels (left panel of a and upper panel of c). (b and lower panel of c) Cell lysates and culture supernatants of HOK-16B cells were subjected to immunoblotting using anti-NLRP10 and anti-β-actin Abs. *P < 0.05 and **P < 0.01 compared with untreated cells.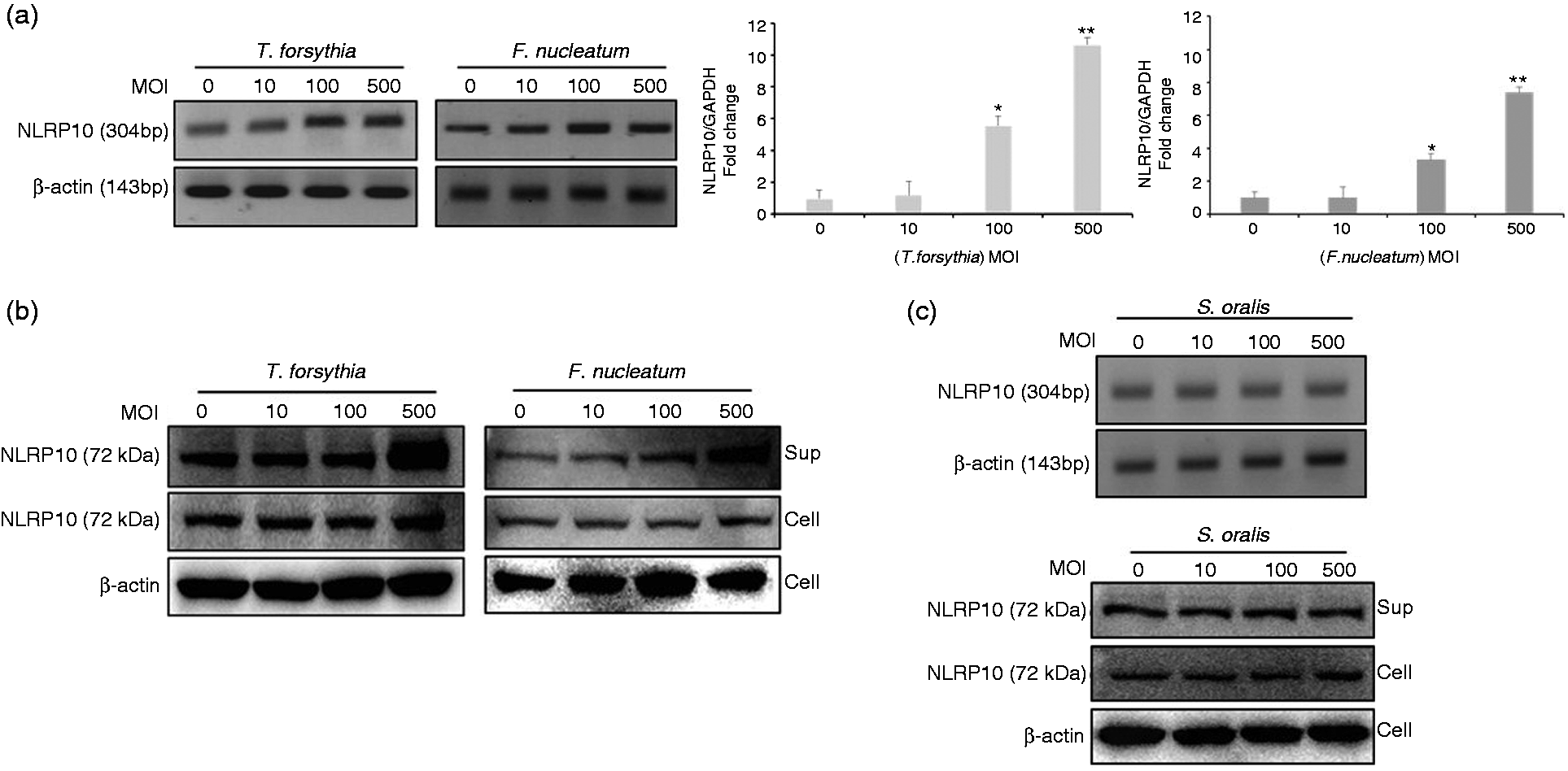
NLRP10 is involved in bacteria-induced IL-1α expression
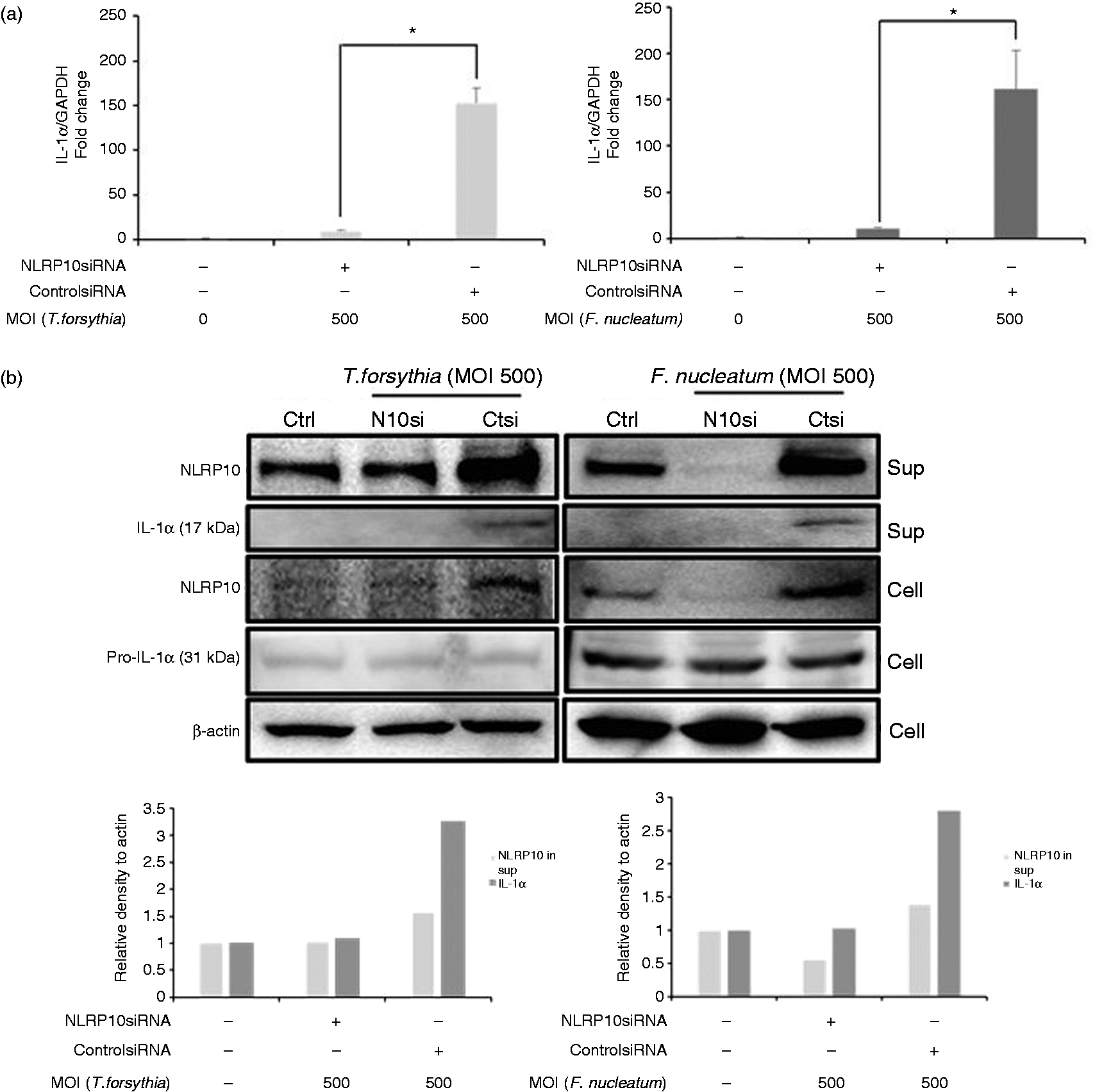
We investigated whether NLRP10 was involved in pro-inflammatory cytokine production. Real-time RT-PCR identified a significant increase in IL-1α mRNA expression in HOK-16B cells infected with T. forsythia and F. nucleatum in a MOI-dependent manner (Figure 2a). We used ELISA to confirm the increase in IL-1α at the protein level (Figure 2b). We determined whether NLRP10 would affect IL-1α expression using siRNA technology. Cells transfected with NLRP10-specific siRNA exhibited significantly lower IL-1α mRNA expression than cells transfected with control siRNA in HOK-16B cells infected with T. forsythia and F. nucleatum (Figure 3a). T. forsythia and F. nucleatum increased COX-2 and IL-8 expression, respectively, and NLRP10 knockdown did not alter this increase (Figure S2). Notably, NLRP10 knockdown remarkably reduced the secreted IL-1α protein, as detected using immunoblotting (Figure 3b). These results suggest that NLRP10 is involved in IL-1α production in HOK-16B cells infected with T. forsythia and F. nucleatum. NLRP10 did not affect bacterial invasion of HOK-16B cells (Figure S3).
IL-1α was induced in HOK-16B cells by T. forsythia and F. nucleatum. HOK-16B cells (5 × 105) were infected with T. forsythia or F. nucleatum at various MOIs for 6 h. (a) The expression of IL-1α mRNA was analysed using real-time RT-PCR. (b) IL-1α levels in culture supernatants were measured using ELISA. The data are shown as the means ± SD of three experiments performed in triplicate. *P < 0.05 and **P < 0.01 compared with untreated cells. NLRP10 knockdown reduced IL-1α expression induced by T. forsythia and F. nucleatum. HOK-16B cells were transfected with NLRP10 siRNA and control siRNA for 24 h, followed by infection with T. forsythia and F. nucleatum for 6 h. (a) The expression of IL-1α mRNA was analysed using real-time RT-PCR. The data are shown as the means ± SD of three experiments performed in triplicate. *P < 0.05 compared with untreated cells. (b) Cell lysates and culture supernatants of HOK-16B cells were subjected to immunoblotting with anti-NLRP10, anti-IL-1α and anti-β-actin Abs. Densitometric data of the blots are shown.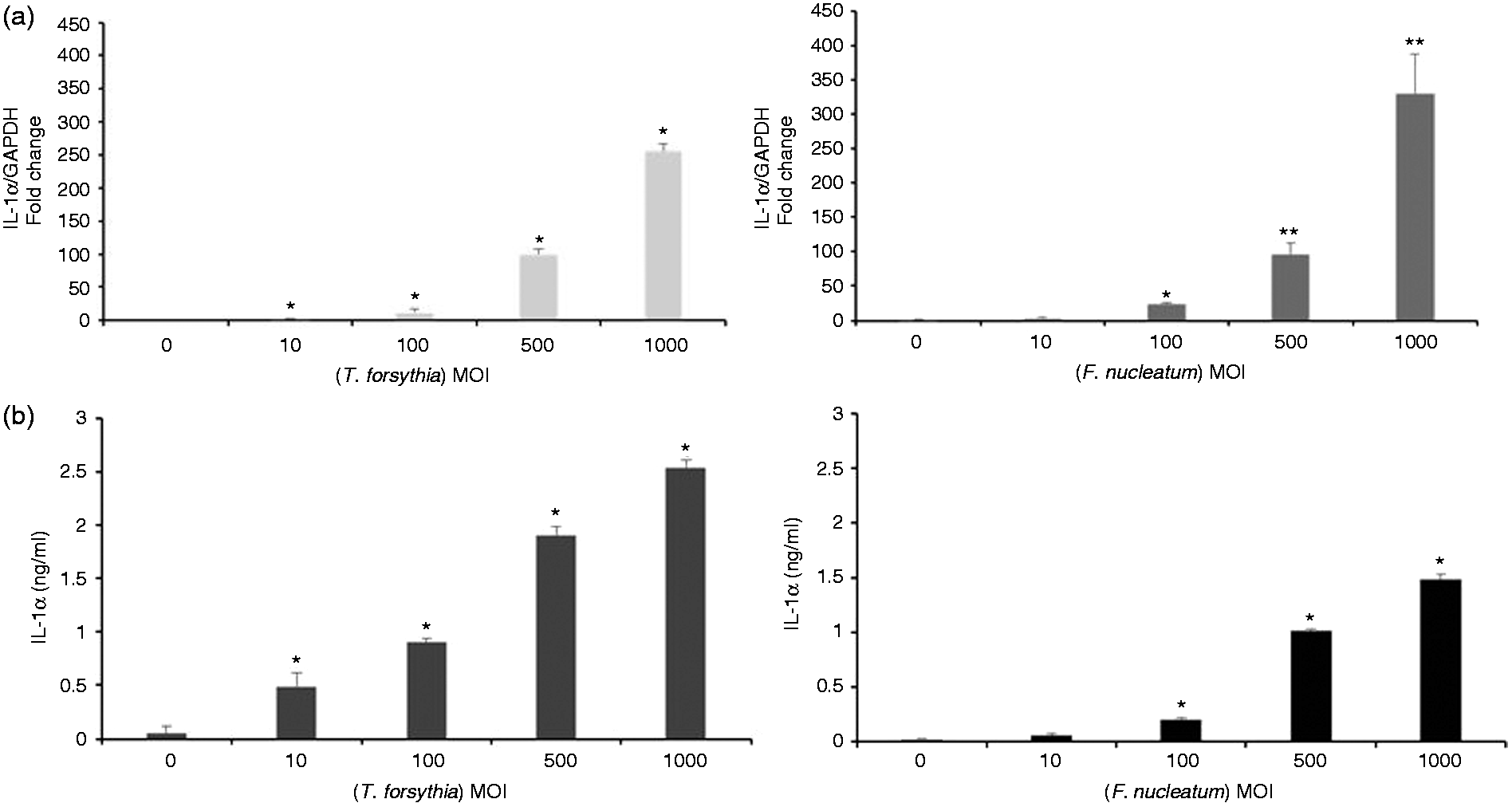
NLRP10 is involved in IL-1α induction via ERK MAPK activation
We analysed the signalling pathway through which NLRP10 regulated IL-1α production. T. forsythia and F. nucleatum activated p38 and ERK MAPKs but minimally activated the SAPK/JNK pathway. The bacteria did not induce IκBα degradation, which indicates that NF-κB signalling was not involved (Figure 4a). NLRP10 knockdown in HOK-16B cells resulted in decreased ERK phosphorylation (P-ERK1/2), but JNK and p38 activation was not affected (Figure 4b). Therefore, we hypothesized that ERK MAPK signalling, as induced by the bacteria, affected IL-1α production. The ERK MAPK inhibitor PD98050 significantly reduced IL-1α mRNA (Figure 4c) and protein levels (Figure 4d). Taken together, these results suggest that NLRP10 is involved in IL-1α production via T. forsythia- and F. nucleatum-induced ERK MAPK activation.
NLRP10 is associated with IL-1α production via ERK activation. (a) HOK-16B cells (5 × 105) and (b) HOK-16B cells (5 × 105) transfected with NLRP10 siRNA and control siRNA were infected with T. forsythia or F. nucleatum for various infection times. Cell lysates were subjected to immunoblotting with anti-IκB, anti-P-ERK, anti-P-SAPK/JNK, anti-P-p38 and anti-β-actin Abs. (c, d) HOK-16B cells (5 × 105) were pretreated with an ERK inhibitor (PD98059) for 30 min and infected with T. forsythia or F. nucleatum for 6 h. The expression of IL-1α mRNA was analysed using real-time RT-PCR. The data are shown as the means ± SD of three experiments performed in triplicate (c). Cell lysates and culture were subjected to immunoblotting with anti-IL-1α and anti-β-actin Abs (d). **P < 0.01 compared with untreated cells.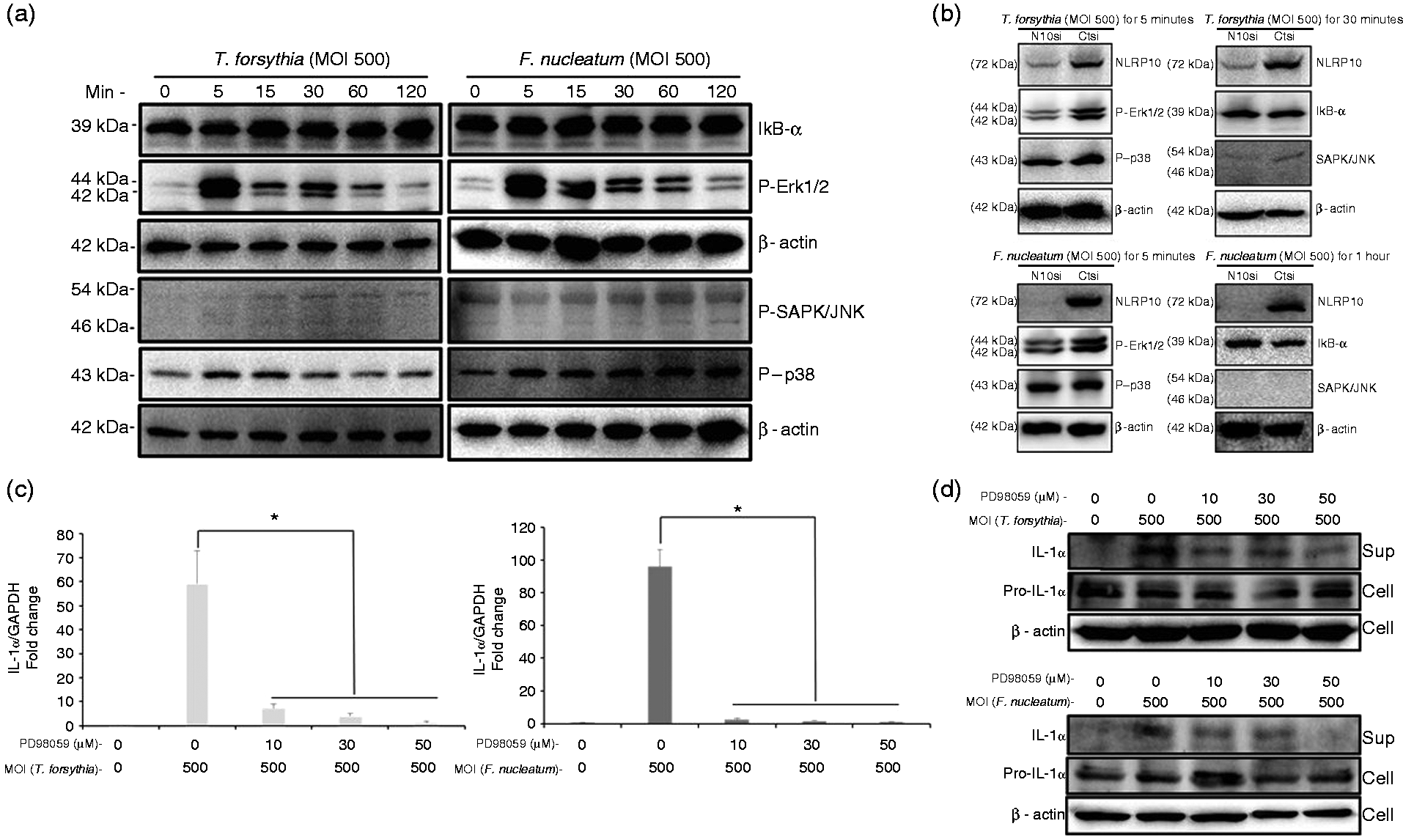
NLRP10 release is partially inhibited by a caspase-1 inhibitor
Notably, we found that NLRP10 release increased in cells infected with periodontal pathogens. We investigated the mechanism of NLRP10 release. Caspase-1 is a well-characterized protease that activates gasdermin D, which forms pores on cellular membranes via oligomerization.26–28 Constitutive caspase-1 activation was observed in HOK-16B cells regardless of bacterial infection (Figure 5a). The caspase-1 inhibitor Ac-YVAD-CHO significantly decreased bacterial infection-induced cell death (Figure 5b) and remarkably reduced bacteria-induced NLRP10 release into the supernatant (Figure 5c). These results suggest that caspase-1 is at least partially involved in NLRP10 release.
NLRP10 release was reduced by a caspase-1 inhibitor. (a, c) HOK-16B cells (5 × 105) were infected with T. forsythia or F. nucleatum for 6 h with (c) or without (a) caspase-1 inhibitor pre-treatment. Cell lysates and culture supernatants were subjected to immunoblotting with anti-caspase-1, anti-NLRP10 and anti-β-actin Abs. Densitometric data of the blots are shown to compare NLRP10 level in supernatants. (b) HOK-16B cells (5 × 104) were infected with T. forsythia or F. nucleatum for 6 h with caspase-1 inhibitor pre-treatment. The presence of LDH released into the culture supernatants was measured using an LDH cytotoxicity assay kit. The data are shown as the mean ± SD of one of three experiments performed in triplicate. **P < 0.01 compared with untreated cells.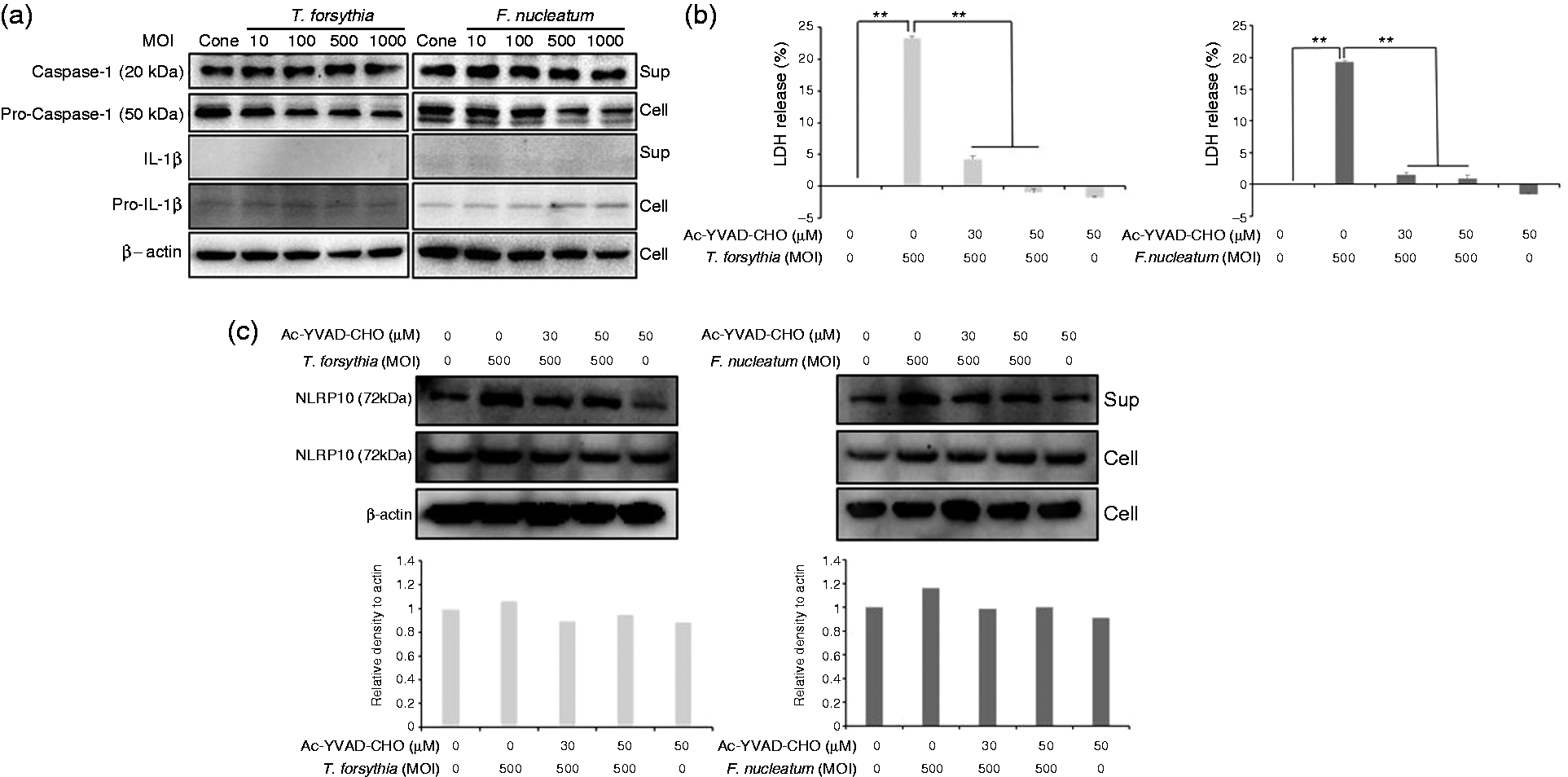
Discussion
This study demonstrated that infection with T. forsythia and F. nucleatum increased NLRP10 expression in HOK-16B cells. This increase was ultimately associated with increased IL-1α expression via ERK activation. The gingival epithelium is in direct contact with bacteria in the subgingival biofilm. The epithelial cells use the innate immune response to create an inflammatory reaction in deeper tissue and combat bacterial infection. IL-1α is a potent pro-inflammatory cytokine that is produced by various cell types. 29 IL-1α promoted epidermal wound healing via stimulation of keratinocyte and fibroblast growth in pig and mouse models.30–32 These characteristics confirm the unique role of IL-1α in maintaining the skin barrier function. However, overexpression of IL-1α may lead to tissue and bone destruction. 33 Prior studies demonstrated that oral epithelial cells infected with F. nucleatum in monospecies and multispecies biofilm exhibited increased IL-1α expression.34,35 Another group found higher levels of IL-1α in gingival crevicular fluid from patients with chronic periodontitis than healthy controls. 36 The IL-1α level positively correlated with the surface area of inflamed periodontal tissue. 36 IL-1α up-regulated RANKL in the human osteoblastic cell line U2OS, 37 and enhanced MMP-9 production in human osteosarcoma cells. 38 We also detected a processed form of IL-1α (17 kDa) in culture supernatants. IL-1α can be proteolytically cleaved at several sites in its N-terminus, which is similar to IL-1β. 39 In contrast to IL-1β, IL-1α is biologically active in its processed and unprocessed forms (31 kDa). However, processed IL-1α has a higher receptor binding affinity and bioactivity than the unprocessed form. IL-1α stimulation increases IL-8 mRNA levels in various cell types, including human keratinocytes.40
NLRP10 inhibits aggregation of apoptosis-associated speck-like protein, which is required for inflammasome activation, resulting in inhibition of caspase-1 activation and IL-1β secretion. 3 Knock-in mice that overexpress NLRP10 are less sensitive to LPS-induced endotoxin than mice with normal NLRP10 expression. 7 NLRP10-deficient mice exhibited increased susceptibility to a systemic C. albicans infection than mice with normal NLRP10 expression. 9 NLRP10 also increased the production of pro-inflammatory cytokines, including IL-6 and IL-8, in HeLa cells and primary human dermal fibroblasts infected with S. flexneri via NF-κB and p38 MAPK activation. 10 T. forsythia and F. nucleatum did not activate NF-κB in our study. IL-8 was produced in HEK 293 T cells infected with F. nucleatum via p38 MAPK activation but without involvement of TLRs, NOD1, NOD2 or NF-κB signalling. 41 F. nucleatum infection in monocytes or macrophages activated NF-κB signalling.42,43 T. forsythia and F. nucleatum activated p38 MAPK in HOK-16B cells, but this activation was not associated with NLRP10 signalling. ERK MAPK was associated with NLRP10, which induced IL-1α production in HOK-16B cells infected with T. forsythia and F. nucleatum. F. nucleatum, a peptidoglycan of F. nucleatum, and outer membrane vesicles of A. actinomycetemcomitans induced NF-κB activation in HEK293T cells transfected with plasmid NOD1 and NF-κB–firefly luciferase reporter plasmid.22,44 Peptidoglycans or components of outer membrane are bacterial ligand candidates for NLRP10 signalling in cooperation with NOD1. Culture supernatants of 10 bacterial species, including F. nucleatum and T. forsythia, regulated the expression of NLRP3 and AIM2 in gingival fibroblasts in an in vitro biofilm model differently according to the protein concentrations of the supernatants. 45 TLR2 and NLRP2 are strongly expressed in HOK-16B cells and their knockdown reduced the expression human β-defensin 3 induced by F. nucleatum. 46 T. forsythia and F. nucleatum may use these PRRs to induce NLRP10 expression. Therefore, elucidation of the PRRs and PAMPs involved in NLRP10 expression may provide insight into NLRP10 signalling. Alternatively, NLRP10 expression may be induced by cytokines that are primarily induced by bacterial infection. TNF-α induced NLRP10 mRNA expression in cerebral microvascular endothelial cells. 47
Notably, we observed increased NLRP10 in culture supernatants, and NLRP10 secretion was partially dependent on caspase-1. Recently, oligomeric NLRP3/ASC inflammasome particles were demonstrated to be extracellularly released and function as danger signals to activate caspase-1. 48 Active caspase-1 cleaves the N-terminal of gasdermin D, which forms pores on cellular membranes.26–28 Caspase-1 was constitutively activated in HOK-16B cells, regardless of bacterial infection in our experimental conditions. However, inhibition of caspase-1 activation partially reduced NLRP10 release. These results indicate that NLRP10 release at least partially requires caspase-1. The role of extracellular NLRP10 must be further elucidated.
In summary, periodontal infection up-regulated NLRP10 expression in HOK-16B cells and augmented pro-inflammatory cytokine IL-1α via ERK signalling. Our data demonstrate the pro-inflammatory role of NLRP10 in HOK-16B cells infected with T. forsythia and F. nucleatum. Therefore, NLRP10 likely plays a critical role in the pathogenesis of periodontitis.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Basic Science Research Program of the National Research Foundation of Korea (NRF-2015R1A2A2A01002598), which was funded by the Ministry of Science, ICT, and Future Planning.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.