
Abstract
Chronic diseases of the respiratory tract, such as cystic fibrosis, are associated with mucosal and systemic hypoxia. Innate immune functions of airway epithelial cells are required to prevent and control infections of the lung parenchyma. The transcription factor hypoxia-inducible factor 1α (HIF-1α) regulates cellular adaptation to low oxygen conditions. Here, we show that hypoxia and HIF-1α regulate innate immune mechanisms of cultured human bronchial epithelial cells (HBECs). Exposure of primary HBECs to hypoxia or the prolyl hydroxylase inhibitor dimethyloxaloylglycine (DMOG) resulted in a significantly decreased expression of inflammatory mediators (IL-6, IFN-γ-induced protein 10) in response to ligands for TLRs (flagellin, polyI:C) and Pseudomonas aeruginosa, whereas the expression of inflammatory mediators was not affected by hypoxia or DMOG in the absence of microbial factors. Small interfering RNA-mediated knockdown of HIF-1α in HBECs and in the bronchial epithelial cell line Calu-3 resulted in increased expression of inflammatory mediators. The inflammatory response was decreased in lungs of mice stimulated with inactivated P. aeruginosa under hypoxia. These data suggest that hypoxia suppresses the innate immune response of airway epithelial cells via HIF-1α.
Introduction
Hypoxia plays an important role in many diseases in terms of aetiology and course. It is known that hypoxia effects on inflammation and inflammation itself leads to hypoxia.1,2 Chronic respiratory diseases are commonly associated with mucosal and systemic hypoxia. In cystic fibrosis (CF) airways, excessive mucus formation generates a hypoxic microenvironment; 3 accordingly, CF lungs are frequently colonized with anaerobic bacteria. 4 However, the role of hypoxia in inflammatory conditions in chronic lung disease remains unclear.
The hypoxia-inducible factor (HIF) regulates cellular adaptation to low oxygen conditions. 5 Human HIF is a heterodimeric transcription factor consisting of an oxygen-sensitive α-subunit with three isoforms (HIF-1α, HIF-2α, HIF-3α) and an oxygen-stable β-subunit (HIF-1β).6,7 Under normoxic conditions, oxygen-dependent hydroxylation of the HIF-α subunit by prolyl hydroxylases results in proteasomal degradation of HIF-α and inactivation of HIF.1,8 Microbial stimuli activate HIF in diverse cells types, including cancer cells, epithelial cells, macrophages and dendritic cells, also under normoxic conditions.9–17 This suggests a distinct function of HIF in innate and adaptive immunity. 18 Myeloid HIF-1α, for instance, is essential in myeloid cell-mediated inflammation and enhances the bactericidal capacity of phagocytes.13,19 In addition, mice deficient in HIF-1α in myeloid cells develop less severe LPS-induced sepsis. 20 HIF-1α also mediates innate immune function of keratinocytes by regulating cathelicidin expression and antibacterial activity. 21 HIF-1α regulates basal defensin expression in intestinal epithelial cells and is protective in murine experimental colitis.22,23
The epithelial surfaces of the respiratory tract are continuously exposed to microbes. Innate and adaptive immune functions of diverse cell types such as resident alveolar macrophages, epithelial cells and dendritic cells are required to prevent and control infections of the lung.24,25 Like professional immune cells, airway epithelial cells are able to detect PAMPs via PRR.25–27 The activation of PRRs results in the release of pro-inflammatory cytokines and increased expression of antimicrobial peptides (AMPs).25–28 In case of infections, the inflammatory response of airway epithelial cells is also increased by macrophage-derived cytokines (e.g. TNF-α, IL-1β), which induce increased surface expression of TLRs and release of inflammatory mediators and AMPs.29–31
The purpose of this study was to characterize the effect of hypoxia on the innate immune response of airway epithelial cells. We hypothesized that hypoxia suppresses the inflammatory response of airway epithelial cells during microbial infection and that HIF-1α mediates the suppressive effects of hypoxia.
Materials and methods
Cell culture
Primary human bronchial epithelial cells (HBECs) and Calu-3 cells were cultured as described previously.32,33 Calu-3 cells were cultured in DMEM/F12 (Life Technologies, Darmstadt, Germany) supplemented with 10% FCS, 100 IU/ml penicillin G and 100 IU/ml streptomycin sulfate. Prior to cell-culture experiments, Calu-3 cells were serum-starved overnight and experiments were carried out in serum-free DMEM/F12. Primary HBECs were cultivated using Airway Epithelial Cell Growth Medium (PromoCell, Heidelberg, Germany). Primary cell-culture experiments presented in this study were performed with cells obtained from six individual patients who underwent surgery because of lung cancer. Only cancer-free tissue was used for cell isolation. The protocol was approved by the institutional review board (ethics committee) of the Landesärztekammer of the State of Saarland, and informed consent was obtained from the patients. Cells were incubated in a hypoxia chamber (2% oxygen, 5% CO2; Coy Laboratory Products, Grass Lake, MI, USA) as indicated. For experiments with polarized epithelial cells primary HBECs grown in air–liquid interface cultures were exposed to hypoxia or normoxia for 3 h and infected with 4 × 103 viable Pseudomonas aeruginosa in 40 µl PBS. 33 To determine the numbers of viable bacteria 6 h after infection, the apical surfaces of the cultures were washed with 100 μl PBS and serial dilutions were plated. Concentrations of IL-6 and IFN-γ-induced protein 10 (IP-10) released into the culture media were measured by ELISA (R&D Systems, Minneapolis, MN, USA). For experiments with dimethyloxaloylglycine (DMOG; Sigma-Aldrich, Hamburg, Germany) cells were pre-incubated with DMOG (0.1 mM and 1 mM) for 3 h before stimulation with microbial stressors. For experiments with CoCl2 cells were pre-incubated with CoCl2 (200 µM) for 3 h before stimulation with microbial stressors without CoCl2. P. aeruginosa strain PAO1 was grown in LB medium and heat-inactivated for 10 min at 95℃. PolyI:C and flagellin were purchased from Invivogen (Toulouse, France). Concentrations of the inflammatory cytokines IL-6 and IP-10 in cell-culture supernatants were assessed by ELISA. Cytotoxicity was determined using an LDH-Cytotoxicity Assay Kit according the manufacturer’s instructions (Abcam, Cambridge, UK).
RNA isolation and real-time RT-PCR
RNA was isolated and reversely transcribed using a RNA isolation Kit (Macherey-Nagel, Düren, Germany) and a cDNA Synthesis Kit (ThermoScientific, Darmstadt, Germany). qRT-PCRs were performed with a SYBR Kit (Bioline, Luckenwalde, Germany) as described before. 32
siRNA experiments
Calu-3 cells or primary HBEC were transfected with siRNA specific for HIF-1α (50 nM; Abcam, Cambridge, UK) using Lipofectamine RNAiMAX (Qiagen, Hilden, Germany) as described before.28,32 Controls were transfected with unspecific control siRNA (50 nM; Abcam) that does not lead to specific degradation of any cellular message. Cells were incubated overnight in medium without antibiotics and treated with polyI:C or flaggelin for 24 h under normoxic conditions as indicated. Supernatants and cell lysates were kept at −80℃.
Mouse experiments
Mice (C57BL/6 N) were maintained under a pathogen-free condition. All animal experiments were approved by the Landesamt für Soziales, Gesundheit und Verbraucherschutz of the State of Saarland following the national guidelines for animal treatment. Mice were kept under normoxia or hypoxia (10% O2 in a ventilated chamber) with O2 concentrations being regulated (autoregulatory unit model 110; Biospherix, Redfield, NY, USA) for 24 h and stimulated intra-nasally with heat-inactivated P. aeruginosa (2 × 107 colony forming units; CFUs) as described before. 32 Twenty-four h after the bacterial stimulation, mice were euthanized, the tracheae were cannulated and a bronchoalveolar lavage (BAL) was performed with 1 ml PBS flushed three times into the lungs. The left lung was homogenated in 1 ml PBS. Total cell numbers and alveolar cells were differentiated as described before. 34 Concentrations of the inflammatory cytokines macrophage inflammatory protein 2 (MIP-2), keratinocyte-derived chemokine (KC) and IL-6 were assessed by ELISA (R&D Systems).
Statistical analysis
Experiments were analysed by t-test (two-sided) or ANOVA (Bonferroni adjustment) for experiments with more than two subgroups. Results were considered statistically significant for P < 0.05. All statistical tests were performed using the software Prism (GraphPad Software, San Diego, CA, USA).
Results
Hypoxia suppresses epithelial expression and release of inflammatory mediators
To determine the effects of hypoxia on innate immune responses of airway epithelial cells primary HBECs were cultured under normoxic or hypoxic (2% O2) conditions for 2 h and stimulated with flagellin (ligand for TLR5), polyI:C (ligand for TLR3) and IL-1β under normoxia or hypoxia for an additional 24 h. Hypoxia did not significantly affect the expression of the pro-inflammatory mediators IL-6 and IP-10 and the AMP hBD-2 by HBECs in the absence of inflammatory stimuli (Figure 1). However, exposure to hypoxia significantly decreased the release of IL-6 by HBECs induced by flagellin (Figure 1a), polyI:C (Figure 1b) and IL-1β (Figure 1c). A robust release of IP-10 by HBECs was only detectable after stimulation with polyI:C, and hypoxia caused a significant decrease in IP-10 release by HBECs incubated with polyI:C (Figure 1b). Furthermore, qRT-PCR showed that hypoxia suppressed the expression of IL-6 (Figure 1d) and hBD-2 (Figure 1e) induced by stimulation with IL-1β, flagellin and polyI:C at a transcriptional level. We further determined whether hypoxia affects the activation of HBECs by P. aeruginosa. Hypoxia significantly decreased the release of IL-6 and IP-10 by HBECs in response to heat-inactivated P. aeruginosa (Figure 2a). Polarized HBECs grown as air–liquid interface cultures were exposed to normoxia or hypoxia and apically infected with viable P. aeruginosa for 6 h. The release of IL-6 and IP-10 (Figure 2b) into the basolateral compartment was significantly decreased under hypoxia after stimulation with viable P. aeruginosa. Moreover, hypoxia resulted in significantly increased numbers of viable P. aeruginosa in washings from the apical surfaces of polarized HBECs 6 h post-infection (Figure 2c).
Hypoxia suppresses the expression and release of inflammatory mediators by airway epithelial cells in response to TLR ligands and IL-1β. Primary HBECs were cultured under normoxic or hypoxic (2% O2) conditions for 2 h and stimulated with (a) flagellin (1 µg/ml), (b) polyI:C (1 µg/ml) and (c) IL-1β (1 ng/ml) under normoxia or hypoxia for an additional 24 h. IL-6 and IP-10 concentrations in supernatants were analysed by ELISA. Epithelial RNA was isolated and qRT-PCR was performed to measure the induction of (d) IL-6 and (e) hBD-2. Data are shown as mean ± SD (n ≥ 3/group). Presented data are representative of three independent experiments. Bars indicate significant differences of *P < 0.05, **P < 0.01 and ***P < 0.001. Hypoxia suppresses the expression and release of inflammatory mediators by airway epithelial cells in response to P. aeruginosa. (a) Primary HBECs were cultured under normoxic or hypoxic (2% O2) conditions for 2 h and stimulated with inactivated P. aeruginosa (P.a.; 107 CFU/ml) under normoxia or hypoxia for additional 24 h. (b, c) Polarized HBECs were cultured under normoxic or hypoxic (2% O2) conditions for 2 h and infected apically with 40 µl viable P. aeruginosa (105 CFU/ml) for 6 h. (a, b) Release of IL-6 and IP-10 was determined by ELISA. (C) The number of viable bacteria was determined in apical washings (100 µl PBS) 6 h post-infection. Data are shown as mean ± SD (n ≥ 3/group). Presented data are representative of two independent experiments. Bars indicate significant differences of **P < 0.01 and ***P < 0.001.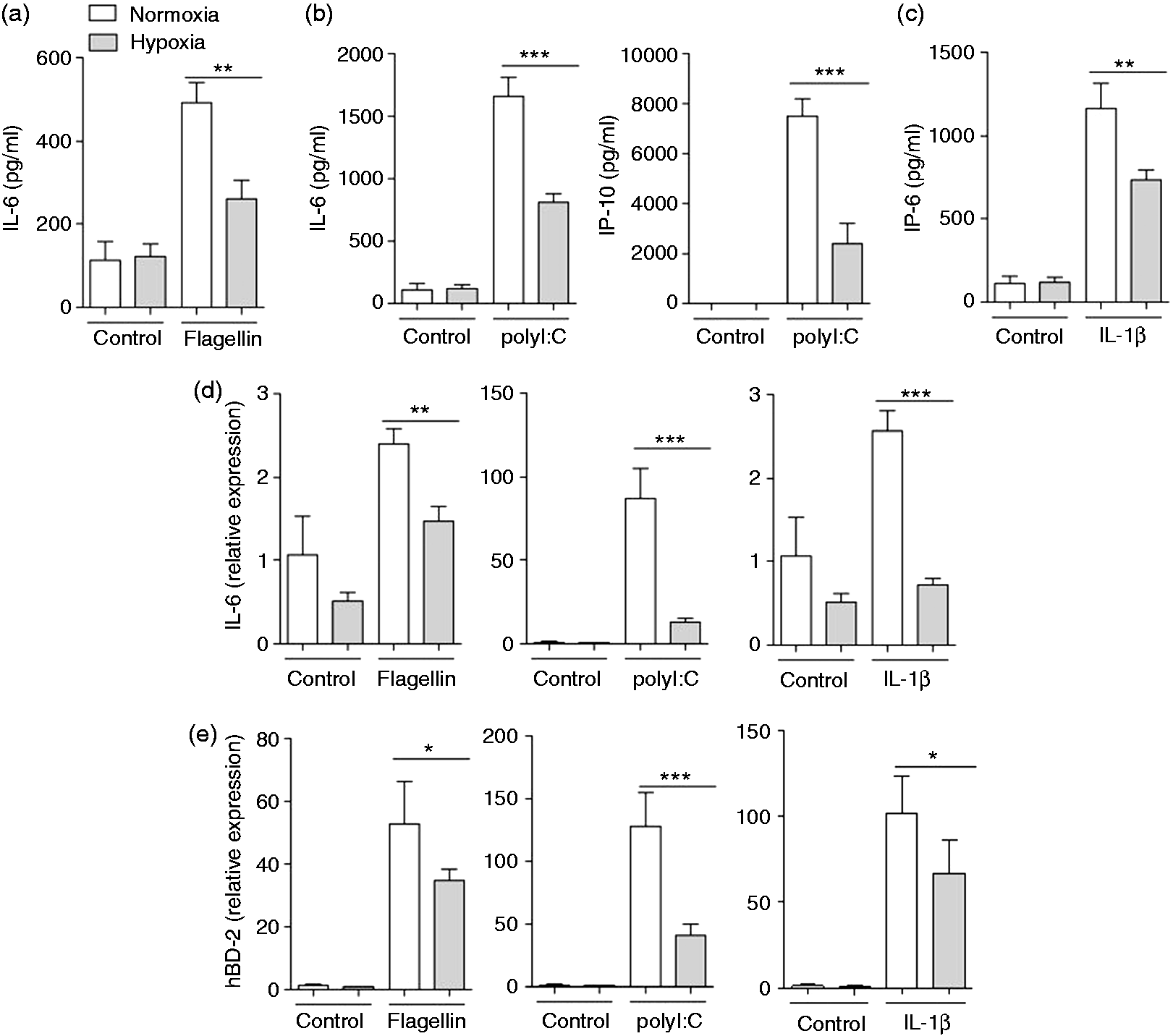
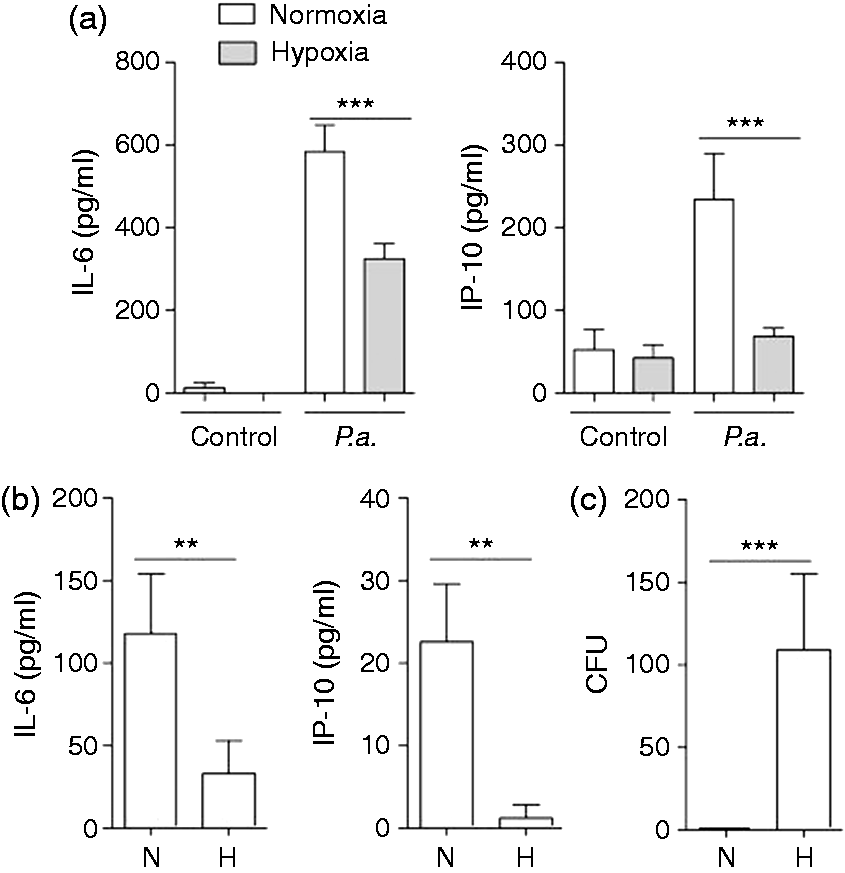
These results demonstrated that hypoxia supresses the inflammatory response of HBECs to microbial infection.
The prolyl hydroxylase inhibitor DMOG and CoCl2 decrease the release of inflammatory cytokines by airway epithelial cells
The prolyl hydroxylase inhibitor DMOG mimics hypoxia by stabilizing HIF-1. To determine whether the effects of hypoxia on the activation of primary HBECs is mirrored by DMOG HBECs were pre-treated with or without DMOG followed by stimulation with polyI:C or heat-inactivated P. aeruginosa in the presence of DMOG. DMOG reduced the release of IL-6 and IP-10 in response to polyI:C (Figure 3a) and P. aeruginosa (Figure 3b). Cobalt chloride (CoCl2) stabilizes HIF-1 and is referred to as hypoxia-mimetic agent.
8
To determine whether the effects of hypoxia on the activation of primary HBECs is mirrored by CoCl2 HBECs were pretreated with or without CoCl2 followed by stimulation with heat-inactivated polyI:C (Figure 3c) and P. aeruginosa (Figure 3d) in media without CoCl2. Pre-treatment with CoCl2 did not affect the release of IL-6 and IP-10 in the absence of P. aeruginosa and polyI:C. However, the release of IL-6 and IP-10 in response to infection with P. aeruginosa and polyI:C was significantly reduced by pre-treatment with CoCl2.
DMOG and CoCl2 decrease the release of inflammatory cytokines by airway epithelial cells. (a, b) Primary HBECs were pre-treated with or without DMOG as indicated followed by stimulation with (a) polyI:C (1 µg/ml) or (b) heat-inactivated P. aeruginosa (P.a.; 107 CFU/ml) in the presence of DMOG. (c, d) Primary HBECs were pre-treated with or without CoCl2 (200 µM) followed by stimulation with (c) polyI:C (1 µg/ml) or (d) heat-inactivated P. aeruginosa (107 CFU/ml) in CoCl2-free media for 24 h. IL-6 and IP-10 concentrations in supernatants were analySed by ELISA. Data are shown as mean ± SD (n ≥ 3/group). Bars indicate significant differences of *P < 0.05 and ***P < 0.001.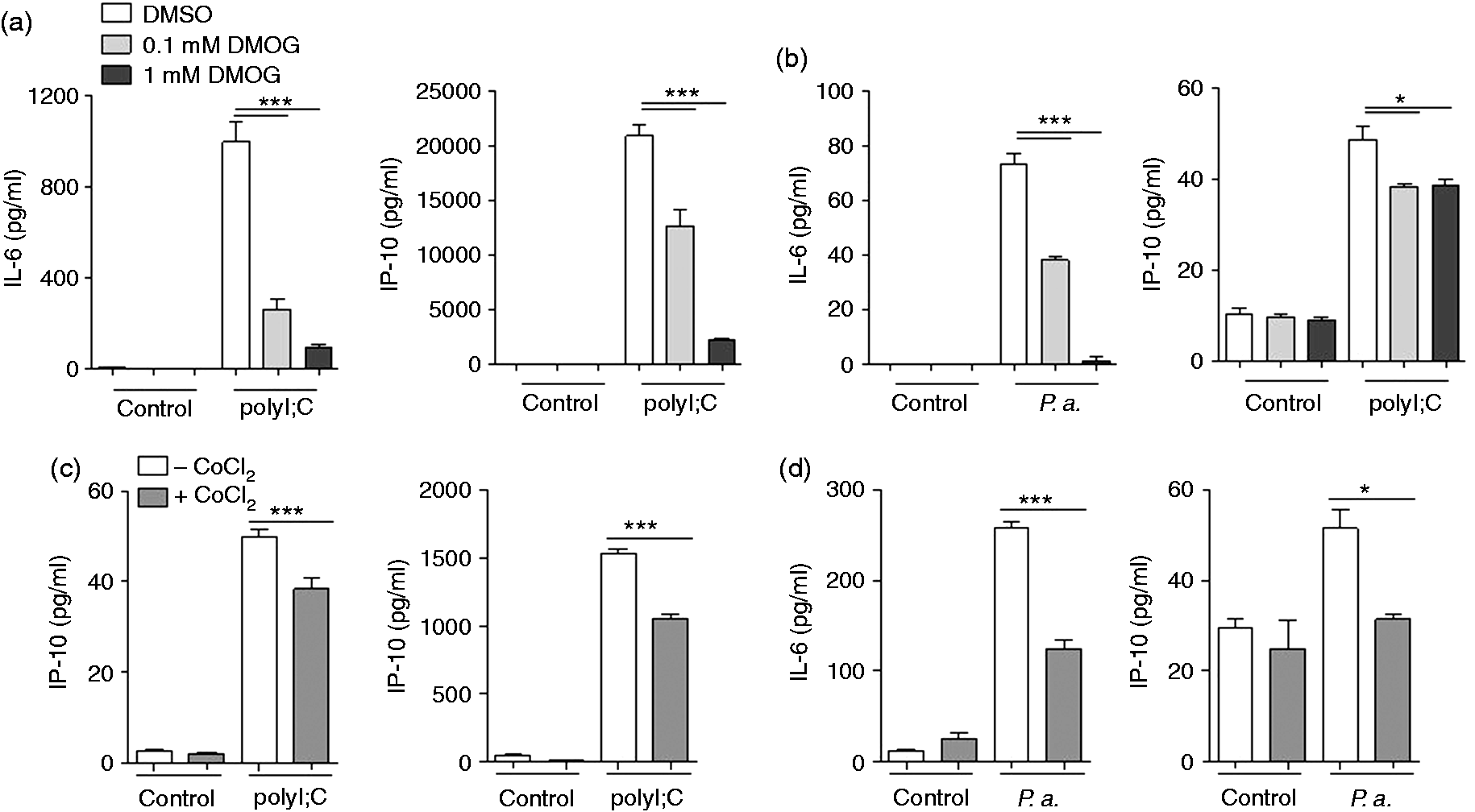
Silencing of HIF-1α leads to increased inflammation in airway epithelial cells
The transcription factor HIF-1α regulates cellular adaptation to low oxygen conditions and host defence mechanisms in various cells types.1,5,19 As hypoxia and DMOG supressed the expression and release of inflammatory mediators in response to microbial stimuli, we next sought to determine whether HIF-1α regulates innate immune responses in airway epithelial cells. Therefore, we silenced HIF-1α in primary HBECs and in the bronchial epithelial cell line Calu-3 via siRNA (Figure 4). HBECs and Calu-3 cells treated with siRNA against HIF-1α or with control siRNA were stimulated with polyI:C or flagellin and cultured under normoxic conditions for 24 h. HIF-1α silencing in HBECs and Calu-3 cells was confirmed by qRT-PCR (Figure 4a). A LDH cytotoxicity assay showed that treatment with siRNA against HIF-1α did not affect viability (Figure 4b). Silencing of HIF-1α increased the basal release of IL-6 and IP-10 by HBECs and Calu-3 cells (Figure 4c–f). Moreover, silencing of HIF-1α increased the release of IL-6 and IP-10 by HBECs and Calu-3 cells in response to stimulation with polyI:C (Figure 4c, d) and flagellin (Figure 4e, f).
Silencing of HIF-1α leads to increased release of inflammatory mediators by respiratory epithelial cells. Primary HBECs and Calu-3 cells were transfected with either unspecific control siRNA (–) or siRNA specific for HIF-1α mRNA (+). (a) Epithelial RNA was isolated and qRT-PCR was performed to measure the expression of HIF-1α. (b) LDH in supernatants as percentage cytotoxicity. (c–f) Twenty-four h after the transfection, cells were cultured under normoxic conditions for 2 h and inoculated with (c, d) polyI:C (1 µg/ml) and (e, f) flagellin under normoxia for an additional 24 h. IL-6 and IP-10 concentrations in supernatants were analysed by ELISA. Data are shown as mean ± SD (n ≥ 3/group). Presented data are representative of two independent experiments. Bars indicate significant differences of *P < 0.05, **P < 0.01 and ***P < 0.001.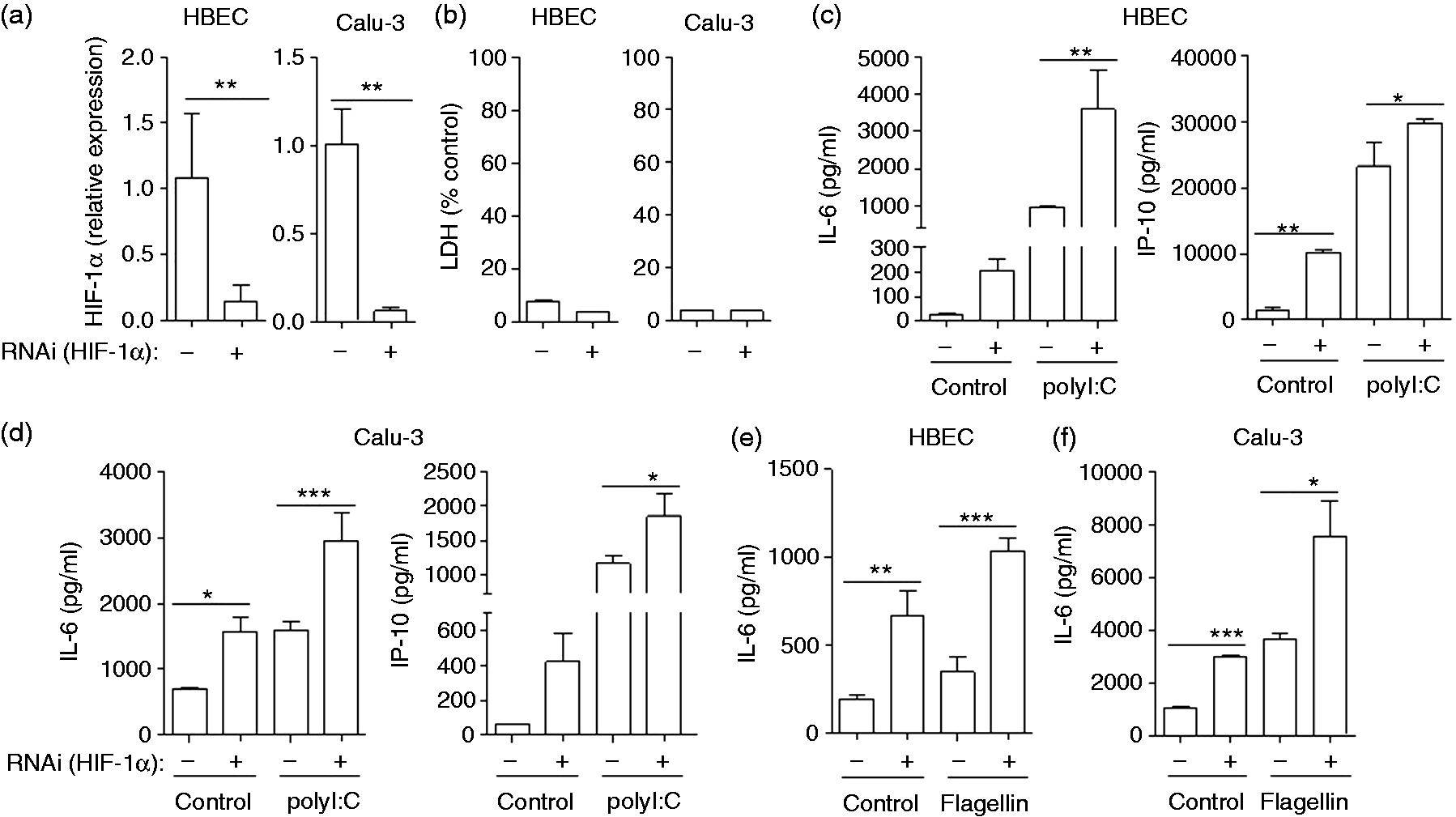
These findings show that siRNA-mediated silencing of HIF-1α in primary HBECs and Calu-3 cells results in an increased release of inflammatory mediators under basal conditions and after stimulation with microbial stressors and suggest that HIF-1α supresses the innate immune responses of airway epithelial cells to microbial stimuli.
Hypoxia reduces inflammation during bacterial lung infection in mice
To evaluate whether hypoxia modulates the inflammatory response to inflammatory stimuli in the lung, mice were kept under hypoxic (10% O2; ‘hypoxic hypoxaemia’) or normoxic conditions for 24 h before infection with heat-inactivated P. aeruginosa for an additional 24 h. Infection with inactivated P. aeruginosa resulted in an inflammation dominated by neutrophils. The number of total immune cells and neutrophils was slightly, but not significantly reduced in mice kept under hypoxic conditions (Figure 5a). Hypoxia resulted in significantly reduced pulmonary levels of inflammatory cytokines. Concentrations of KC (Figure 5b), MIP-2 (Figure 5c) and IL-6 (Figure 5d) were significantly reduced in the BAL fluids and lung tissue of hypoxic mice compared with normoxic mice.
Concentrations of inflammatory cytokines are reduced in lungs of mice exposed to hypoxia. Mice were exposed to hypoxia (10% O2) or normoxia for 24 h, stimulated intra-nasally with heat-inactivated P. aeruginosa in PBS, and exposed to hypoxia or normoxia for an additional 24 h. (a) Numbers of total immune cells, neutrophils and macrophages were determined in BAL fluids. Concentrations of (b) KC, (c) MIP-2 and (d) IL-6 were measured in BAL fluids and in lung homogenates. Data are shown as mean SEM (n ≥ 7/group). Bars indicate significant differences of *P < 0.05 and ***P < 0.001.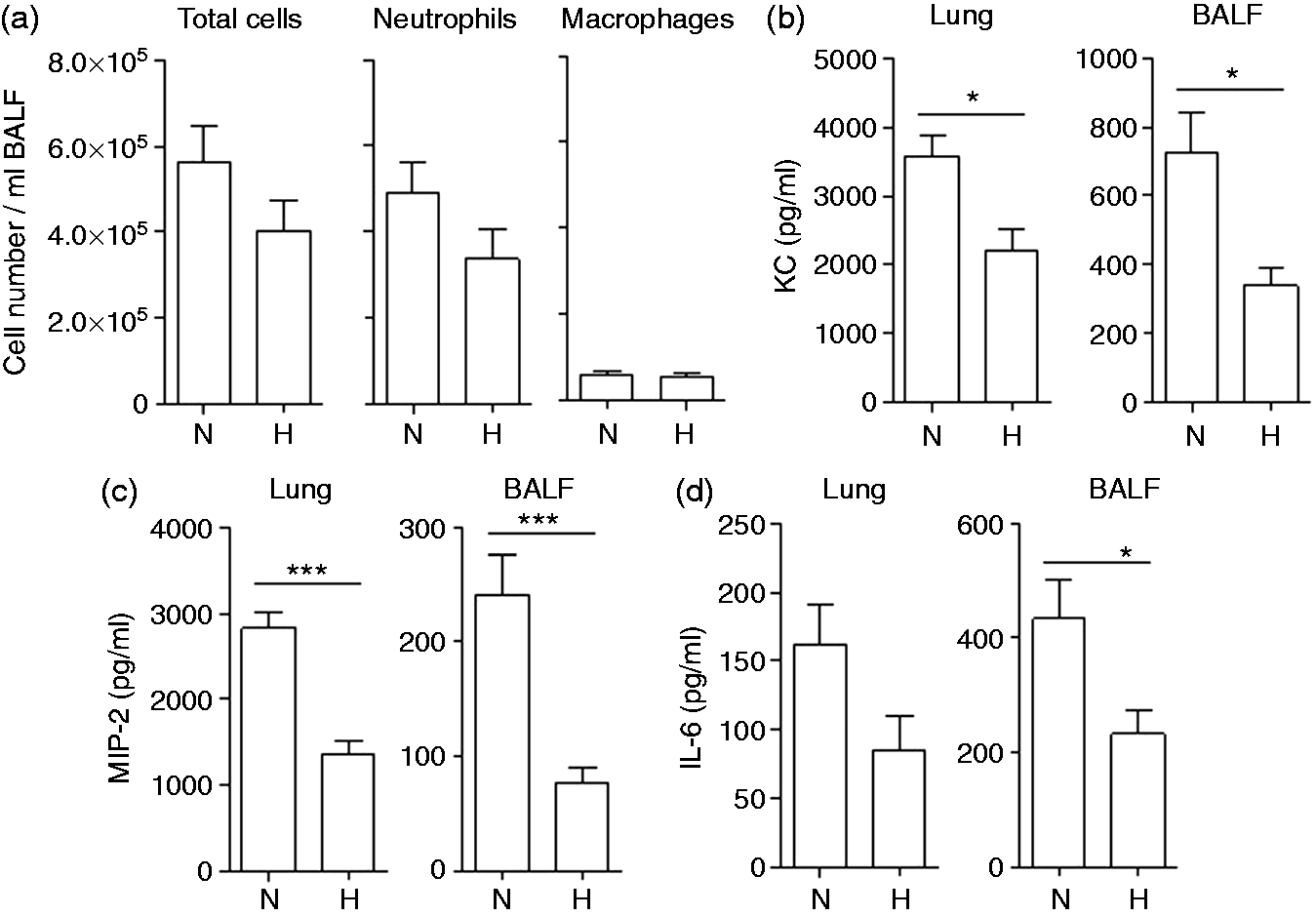
Discussion
In this study, we analysed the consequence of hypoxia in the host defence of the respiratory epithelium. The main finding is that hypoxia suppresses innate immune functions of the airway epithelial cells during microbial infection. We have shown that exposure to hypoxia impairs the inflammatory response of airway epithelial cells. Studies with hypoxia-mimetic agents (DMOG, CoCl2) and RNAi indicate that the effects of hypoxia on airway epithelial cells are largely mediated by HIF-1α.
Patients with chronic respiratory tract diseases are often hypoxic and infections of the lung are frequently associated with local and systemic hypoxia.3,35,36 Hypoxaemia, for instance, is common in acute exacerbations in patients with chronic obstructive pulmonary disease. 37 Mucosal hypoxia was also detected at the epithelial surfaces of patients with CF, which are frequently infected with P. aeruginosa. 3 Our finding that hypoxia suppresses innate immune functions of the respiratory epithelium during microbial infection suggests that a hypoxic micromilieu contributes to microbial infection of mucosal lung surfaces in patients with chronic respiratory tract disease. Remarkably, existing hypoxia affected innate immune functions of respiratory epithelial cells stimulated with P. aeruginosa, bacterial flagellin and polyI:C, a synthetic analogue of double-stranded viral RNA. Thus, low oxygen concentrations seem to favour bacterial colonization, as well as viral infections of the mucosal surfaces of the lung. Our data also suggest that decreased expression of epithelial antimicrobial peptides directly contributes to enhanced bacterial growth under hypoxic conditions. Suppressed release of epithelial-derived chemokines may further contribute to an insufficient influx and activation of immune cells during initial microbial infection.
Our finding that hypoxia and CoCl2 decrease and knock-down of HIF-1α increases inflammation in respiratory epithelial cells is rather surprising as HIF-1α is described as a master regulator of innate immunity in professional immune cells by promoting inflammation and innate immune mechanisms.13,19,38 HIF-1α regulates, for instance, myeloid cell metabolism, inflammation, invasiveness and bactericidal capacity of phagocytes.13,19 Mice deficient in HIF-1α in the myeloid lineage also develop less severe LPS-induced sepsis likely due to a reduced expression of inflammatory cytokines such as TNF-α released by macrophages. 20 Thus, low oxygen levels, which are often found in infected tissues, and microbial factors enhance innate immune functions of immune cells at the site of infection via HIF-1α.13,19,38 However, the effect of hypoxia in cellular functions appears to be cell-type specific. Hypoxia, for instance, decreases the expression of receptors of the innate immune system (e.g. TLR4, TNF cell surface receptor 1) and the expression of IL-8 in cultured endothelial cells.39,40 In a murine colitis model, decreased epithelial HIF-1 expression correlated with more severe clinical symptoms, and constitutive activation of HIF displayed increased expression levels of HIF-1-regulated barrier-protective genes resulting in attenuated loss of barrier. 23 Hypoxia strongly attenuates internalization of the P. aeruginosa into alveolar and bronchial epithelial cells, 41 and activation of HIF-1 also protects bronchial epithelial cells against oxidant-induced loss of barrier integrity. 42 Moreover, Eckle et al. 43 showed that HIF-1α is protective in acute lung injury by optimizing carbohydrate metabolism in the alveolar epithelium resulting in decreased inflammation. 43 The experiments presented in this study with specific ligands for TLRs (polyI:C for TLR3 and flagellin for TLR5) showed that hypoxia directly suppresses the activation of mucosal immunity and inflammation via TLR-dependent signalling cascades. Even although additional studies are required to uncover the exact cellular mechanism behind the impact of hypoxia on TLR-signalling, our experiments with the hypoxia-mimetic agent CoCl2, which activates HIF-1, 8 and siRNA directed against HIF-1α indicate that HIF-1 mediates the suppressive effects of hypoxia on epithelial innate immunity. Our data are in line with the study of Eckle et al., 43 which also showed that HIF-1α repression results in increased release of markers for epithelial cell inflammation in the bronchial epithelial cell line Calu-3 exposed to stretch. Thus, HIF-1α-regulated carbohydrate metabolism could contribute to the attenuated inflammatory response of respiratory epithelial cells to microbial under hypoxia in our in vitro models.
Our study has several limitations. In our experiments, we used an oxygen fraction of 0.21 as normoxic control. This might be higher than the oxygen fraction which the bronchial epithelium is exposed to in vivo, as the alveolar oxygen fraction is only 0.14. However, to our knowledge all available literature about the impact of hypoxia on lung epithelial cells defined 0.21 as normoxia.13,41,44–47 Hence, our current data are comparable with those in the established literature. In addition, our study did not aim to investigate the exact mechanism by which HIF-1α regulates innate immune mechanisms in bronchial epithelial cells. Possible mechanisms could include changes in carbohydrate metabolism, as described by Eckle et al. 43
In conclusion, we show that hypoxia suppresses the innate immune response and inflammation of airway epithelial cells via HIF-1α. Our data suggest that hypoxia not only favours microbial infection of mucosal surfaces of the lung, but also counteracts tissue damage during infection by dampening inflammation. Thus, HIF-1α in airway epithelial cells exerts a tissue specific role in the regulation of host immunity.
Footnotes
Acknowledgements
We thank Anja Honecker for excellent technical assistance.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants from DFG BE 4813/1-1 to CB and the Federal Ministry of Education and Research (FKZ 01GI0881-0888) to RB.