
Abstract
Myeloid cells such as macrophages are critical to innate defense against infection. IL-1 receptor-associated kinase M (IRAK-M) is a negative regulator of TLR signaling during bacterial infection, but the role of myeloid cell IRAK-M in bacterial infection is unclear. Our goal was to generate a novel conditional knockout mouse model to define the role of myeloid cell IRAK-M during bacterial infection. Myeloid cell-specific IRAK-M knockout mice were generated by crossing IRAK-M floxed mice with LysM–Cre knock-in mice. The resulting LysM–Cre+/IRAK-Mfl/wt and control (LysM–Cre–/IRAK-Mfl/wt) mice were intranasally infected with Pseudomonas aeruginosa (PA). IRAK-M deletion, inflammation, myeloperoxidase (MPO) activity and PA load were measured in leukocytes, bronchoalveolar lavage (BAL) fluid and lungs. PA killing assay with BAL fluid was performed to determine mechanisms of IRAK-M-mediated host defense. IRAK-M mRNA and protein levels in alveolar and lung macrophages were significantly reduced in LysM–Cre+/IRAK-Mfl/wt mice compared with control mice. Following PA infection, LysM–Cre+/IRAK-Mfl/wt mice have enhanced lung neutrophilic inflammation, including MPO activity, but reduced PA load. The increased lung MPO activity in LysM–Cre+/IRAK-Mfl/wt mouse BAL fluid reduced PA load. Generation of IRAK-M conditional knockout mice will enable investigators to determine precisely the function of IRAK-M in myeloid cells and other types of cells during infection and inflammation.
Introduction
Respiratory bacterial [e.g. Pseudomonas aeruginosa (PA)] infections are involved in the pathogenesis of various lung diseases, including chronic obstructive pulmonary diseases and cystic fibrosis (CF).1–3 Initiation and resolution of lung innate immune/inflammatory responses, including recruitment and activation of leukocytes of myeloid lineage (e.g. macrophages and neutrophils), are critical to contain bacterial infections appropriately.4,5 Excessive inflammation or activation of myeloid cells is detrimental to patients suffering from various lung diseases.6,7 Thus, a balanced lung pro- and anti-inflammatory response is essential to maintain a normal host defense mechanism.
IL-1 receptor-associated kinase M (IRAK-M), also known as IRAK-3, is primarily expressed in monocytes and macrophages, and serves as a negative regulator of TLR signaling in response to bacterial infection.8,9 It is widely recognized that myeloid cells such as macrophages are critical to maintaining lung homeostasis during infection.10,11 Previous studies using whole-body IRAK-M knockout mice have demonstrated the inhibitory role of IRAK-M in lung pro-inflammatory response to PA infection.12,13 Moreover, previous in vitro studies have shown that isolated lung macrophages from IRAK-M knockout mice were more pro-inflammatory in response to LPS stimulation. However, the in vivo role of myeloid cell IRAK-M in bacterial infection is unclear. To define clearly the contribution of myeloid cell IRAK-M in lung response to bacterial infection, we have generated a novel myeloid cell-specific IRAK-M deletion mouse model by using the cre-loxP system, and evaluated functional consequences of myeloid cell IRAK-M deletion in a murine model of PA infection.
Material and methods
Ethics statement
Experimental animals used in this study were covered by a protocol approved by Institutional Animal Care and Use Committee of National Jewish Health. All mice were bred and housed in our Biological Resource Center under pathogen-free conditions, and tested to establish that they were virus- and Mycoplasma pulmonis-free. All experimental procedures were carried out to minimize animal discomfort, distress and pain, by following the American Veterinary Medical Association Guidelines.
Generation of mice with IRAK-M deletion in myeloid cells
Generation of IRAK-M-floxed mice
IRAK-M gene targeted (floxed) C57BL/6 embryonic stem (ES) cell lines (JM8.N4) were purchased from European Conditional Mouse Mutagenesis Program (EUCOMM), (Munich-Neuherberg, Germany). As shown in Figure 1, the targeting construct is a knockout first conditional second design, where the LacZ gene serves both to disrupt the gene and act as a reporter of gene expression in the original targeted form. Subsequent removal of LacZ and the neomycin selection cassette by Flp recombinase-driven excision converts the allele to a conditional knockout where exon 3 of the IRAK-M gene is floxed. To create chimeric founder mice (F0), the IRAK-M-floxed ES cells were expanded and microinjected into albino B6 (C57BL/6BrdCrHsd-Tyrc) mouse blastocysts at National Jewish Health’s Mouse Genetics Core Facility. The blastocysts were then transferred into pseudopregnant female recipients (outbred ICR mice). Chimeric male mice (F0) were then bred to albino C57BL/6 (C57BL/6BrdCrHsd-Tyrc) females to establish germline transmission. F1 pups with black coat color were screened for the targeted allele by genotyping using PCR with IRAK-M 5’ forward primer CTTTCGTGAGACACAACACAGAGC and IRAK-M 3’ reverse primer AACTGGAATCGATACATTCACACC. Then, these mice were bred to FLPO recombinase driver mice [B6-tyrjC57BL/6N-Tg(CAG-FlpO)1Afst/Mmucd], generously provided by the University of Michigan Transgenic Core and derived by crossing the C57BL/6N-Tg(CAG-Flpo)1Afst/Mmucd mice from the MMRRC to albino C57BL/6 mice+ to induce excision of the LacZ-neomycin cassette and convert the IRAK-M modification to a conditional allele. Genotyping by PCR identified mice where the LacZ-neomycin cassette had been excised from the floxed IRAK-M allele. These mice were then bred together to create IRAK-Mfl/fl mice lacking the FlpO transgene.
Strategy for generating IRAK-M conditional knockout mice. After sequential crossing of mice carrying a modified IRAK-M allele, first to FlpO recombinase driver mice, and then to LysM–Cre knockin mice, the allele was converted to the floxed allele where exon 3 of IRAK-M was successfully deleted in myeloid cells (e.g. macrophages).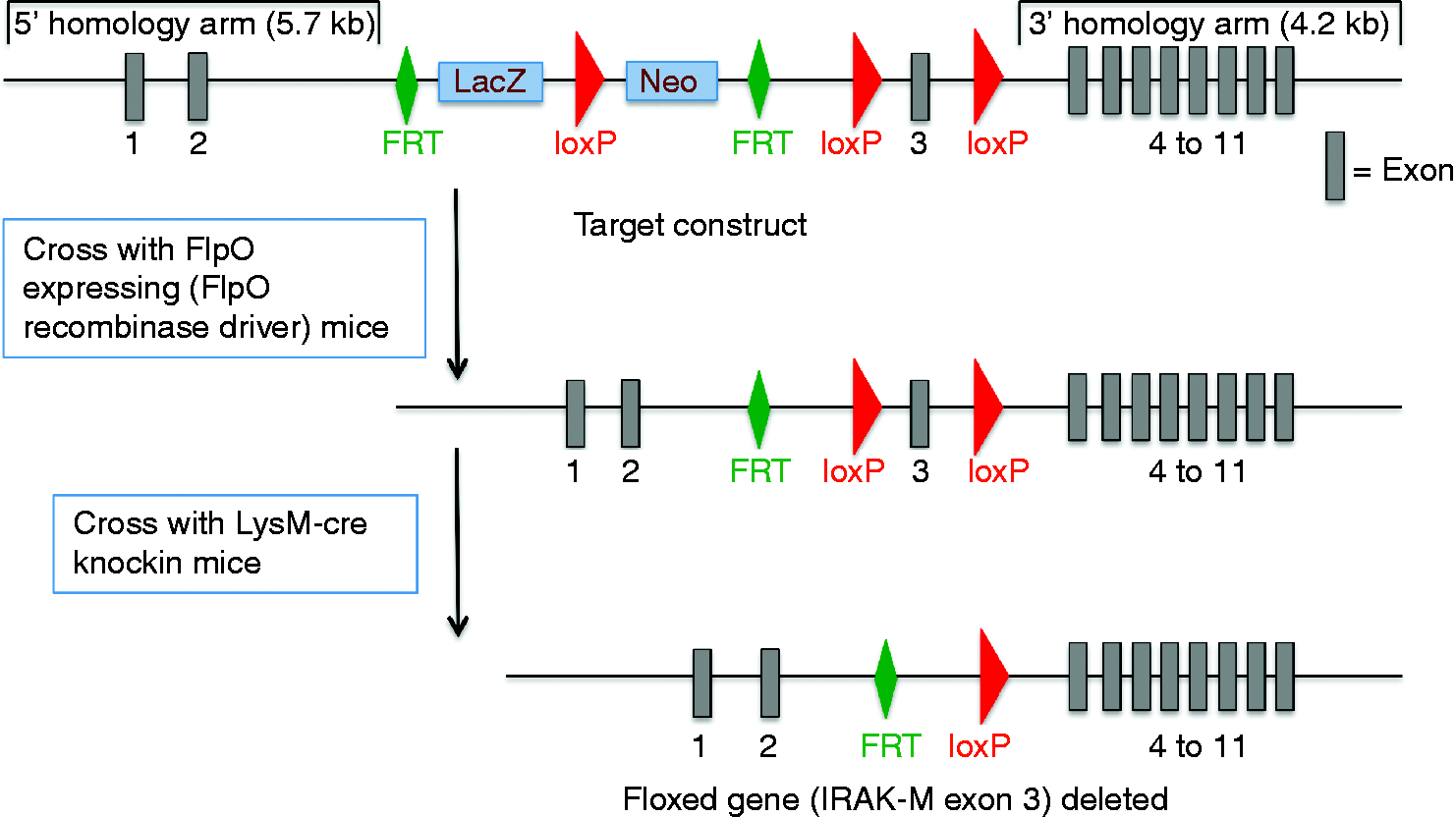
Generation of homozygous IRAK-M floxed mice (F3)
We generated homozygous IRAK-M floxed mice by crossing 8-wk old heterozygous IRAK-Mfl/fl females and males. All of the F3 pups were genotyped by PCR to confirm homozygous for carrying floxed IRAK-M gene (Figure 2a).
Confirmation of IRAK-M floxed allele and cre expression by genotyping. DNA extracted from mouse ear clips was used for gel-based PCR to identify (a) IRAK-M floxed allele and (b) Cre expression. Lanes 1, 3 and 5 = LysM–Cre+/IRAK-Mfl/wt mice. Lanes 2 and 4 = LysM–Cre–/IRAK-Mfl/wt mice.
Breeding for generation of myeloid cell-specific conditional IRAK-M deletion mice
LysM–Cre knock-in mice [B6.129P2-Lyz2tm1(cre)Ifo/J, stock# 004781] were purchased from Jackson Laboratory (Bar Harbor, ME, USA). In these mice, Cre recombinase has been introduced into the LysM locus. LysM is expressed in cells of the myeloid cell lineage, including monocytes, mature macrophages and granulocytes. To generate mice that lack IRAK-M expression specifically in myeloid cells, homozygous floxed IRAK-M female mice were crossbred with LysM–Cre male mice (C57/BL6 background) to produce heterozygous IRAK-M floxed mice with LysM–Cre expression (LysM–Cre+/IRAK-Mfl/wt). To generate the control mice under the same genetic background, homozygous floxed IRAK-M female mice were crossbred with C57/BL6 (LysM–Cre negative) male mice to produce heterozygous IRAK-M floxed mice without LysM–Cre expression. We were not able to generate homozygous myeloid cell IRAK-M-deleted mice because the IRAK-M and LysM genes are positioned in close proximity (roughly 3000 kb apart) on chromosome 10. As such, the possibility of achieving a crossover event between these two genes so that a mouse could be modified at both the LysM and the IRAK-M loci is very remote and something that we did not observe in all of the breeding and genotyping that we performed. Therefore, only heterozygous myeloid cell IRAK-M-deficient mice (LysM–Cre+/IRAK-Mfl/wt) were available for our study. All mice were genotyped for the IRAK-M floxed band at 540 bp and/or wild type band at 407 bp, as well as the band for cre expression at 250 bp by PCR prior to be used in our study (Figure 2b).
Preparation of PA
We used a CF clinical isolate of mucoid PA for our mouse study as previously described. 14 Briefly, PA was first streaked onto a tryptic soy agar plate and cultured for 18–22 h at 37℃. An individual colony was then inoculated into tryptic soy broth and shaken at 225 rpm at 37℃ to grow the bacteria until 1 × 109 CFUs/ml were achieved as determined by spectrophotometry (OD at 600 nm = 1.0).
Mouse model of PA infection
LysM–Cre+/IRAK-Mfl/wt and LysM–Cre–/IRAK-Mfl/wt mice were intranasally infected with PA at 107 CFUs/mouse. After 24 h of infection, mouse lungs were lavaged with 1 ml sterile saline. Cell-free bronchoalveolar lavage (BAL) fluid was stored at –80℃ for cytokine analysis. Cytospins of BAL cells were stained with a Diff-Quick Kit (IMEB INC., San Marcos, CA, USA), and leukocyte differentials were determined as percentage of 500 counted leukocytes. The left lung lobes from infected mice were homogenized in PBS, and then cultured on tryptic soy agar plates to quantify PA levels.
Mouse lung histological analysis
The apical lobe of the mouse right lung was fixed in 10% formalin, embedded in paraffin and cut at 5 µm thickness for histological analysis. Hematoxylin and eosin-stained lung sections were evaluated in a blinded fashion under the light microscope using a histopathologic inflammatory scoring system, as previously described. 15 A final score per mouse on a scale of 0 to 26 (least to most severe) was obtained based on an assessment of the quantity and quality of peribronchiolar and peribronchial inflammatory infiltrates, luminal exudates, perivascular infiltrates and parenchymal pneumonia.
Lung tissue myeloperoxidase activity assay
Myeloperoxidase (MPO) is mainly released by neutrophils and has been used as an index of tissue neutrophil activation levels. MPO activity levels in BAL fluid were measured to reflect neutrophil activity as we previously described. 16 Briefly, BAL fluid was collected after centrifugation at 20,817 g for 5 min. The assay solution was prepared fresh by mixing a 50 mM potassium phosphate buffer (pH 6.0) containing 0.167 mg/ml o-dianisidine dihydrochloride and 0.0005% hydrogen peroxide (Sigma-Aldrich, St. Louis, MO, USA). Cell-free BAL fluid (100 µl) was added to a 96-well plate. Then, 200 µl of the assay solution was added and mixed rapidly. The enzymatic activity was determined spectrophotometrically by measuring the change in absorbance at 405 nm using a plate reader. MPO activity was expressed as an OD value.
Assay for MPO-mediated killing of PA
Sterile filtered BAL fluid was pre-treated with ABAH (20 nM; Sigma-Aldrich), a potent MPO inhibitor, or DMSO (as control) for 10 min, and then added to PA in a 1.5-ml tube (103 CFUs/ml) to incubate for 2 h at 23℃. PA load was quantified by culturing the bacteria on agar plates. This assay allowed us to test whether extracellular MPO in BAL fluid has a direct bactericidal effect.
Confirmation of IRAK-M deletion in mouse primary alveolar macrophages
Primary alveolar macrophages (pAMs) were isolated from BAL cells of untreated LysM–Cre+/IRAK-Mfl/wt and LysM–Cre–/IRAK-Mfl/wt mice to measure IRAK-M expression. BAL cells were collected, spun down, re-suspended in RPMI 1640 medium with 10% FBS and seeded in 48-well culture plates (4 × 104 cells per well) for 2 h to allow cells to adhere. Thereafter, non-adherent cells were washed off and removed. Adherent cells (pAMs) were harvested in RNA lysis buffer to evaluate IRAK-M and cre mRNA levels.
Confirmation of IRAK-M deletion in mouse lung macrophages
Lung macrophages were isolated to confirm IRAK-M protein deletion by Western blot. Left lungs from individual mice were collected, cut into small pieces and digested with collagenase/dispase buffer (Sigma-Aldrich) for 75 min at 37℃. After the digestion, 0.6 mM EDTA was added to incubate for another 10 min at 37℃. The lung tissue/supernatant mixture was then passed through a sterile Cellector tissue sieve (Fisher Scientific, Waltham, MA, USA) to obtain a single-cell suspension. The collected cells were treated with red cell lysis buffer (Fisher Scientific), spun down, re-suspended in RPMI 1640 medium with 10% FBS and seeded in 12-well culture plates (4 × 104 cells per well) for 2 h to allow cells to adhere. Adherent lung macrophages were harvested in RIPA lysis buffer (Fisher Scientific) to evaluate lung IRAK-M protein expression.
Western blot analysis of BAL and lung macrophage IRAK-M
Equal amount cell lysate proteins was separated by SDS-PAGE, transferred onto PVDF membranes and probed with rabbit anti-IRAK-M (EMD Millipore, Billerica, MA, USA) or mouse anti-GAPDH (Santa Cruz Biotechnology, Dallas, TX, USA) Ab. Densitometry was performed to quantify proteins.
Collection of mouse blood leukocytes
Blood from mice was collected by cardiac puncture. After lysis of red blood cells, leukocytes were spun down by centrifugation and saved in RLT lysis buffer for total RNA extraction.
Culture of mouse primary tracheal epithelial cells
We used airway epithelial cells as a negative control to further confirm the specificity of IRAK-M deletion in myeloid cells. We are aware of IRAK-M is primarily expressed in monocytes and macrophages, but previous publications have shown epithelial cells express IRAK-M as well.17,18 Tracheas from the non-infected LysM–Cre+/IRAK-Mfl/wt and LysM–Cre–/IRAK-Mfl/wt (control) mice were isolated by digesting individual trachea with 0.1% protease at 4℃ for 16 h. The released cells were seeded on collagen-coated polyester transwell inserts (12-well plate, 0.4 µm pore size; Corning Costar, Corning, NY, USA) at 4 × 104 cells in 500 µl DMEM/BEBM (1:1) with supplements19,20 Cells were grown in immersed culture for 7 d to obtain pure epithelial population and then harvested in RLT lysis buffer to evaluate cre and IRAK-M mRNA expression.
ELISA of mouse KC and TNF-α
KC, a chemokine for neutrophils, was quantified in mouse BAL fluid, and TNF-α was measured in pAM with or without LPS (100 ng/ml) stimulation for 24 h by using a mouse KC or TNF-α DuoSet ELISA Development kit (R&D Systems, Minneapolis, MN, USA), as per the manufacturer’s instruction.
Quantitative real-time RT-PCR
TaqMan gene expression assay for exon 3 of mouse IRAK-M gene was designed as follows: forward primer 5’- GCATCAACGAGCTATCCACT -3’; reverse primer 5’- TTCCAGAGGTCC-AGGGTC -3’; probe 5’- TTCCATTCCCCAGCTTCCCACC -3’. Quantitative real-time PCR was performed on a CFX96 real-time detection system (Bio-Rad Laboratories, Hercules, CA, USA). The housekeeping gene 18sRNA was evaluated as an internal positive control to normalize IRAK-M mRNA level. The comparative threshold cycle method (ΔΔCT) was used to demonstrate relative IRAK-M mRNA levels.21,22
Statistical analysis
Data are presented as mean ± SEM. One-way ANOVA was used for multiple comparisons, and Tukey’s post hoc test was applied where appropriate. Student’s t-test was used when only two groups were compared. A P-value < 0.05 was considered significant.
Results
Confirmation of cre-mediated myeloid cell-specific IRAK-M deletion
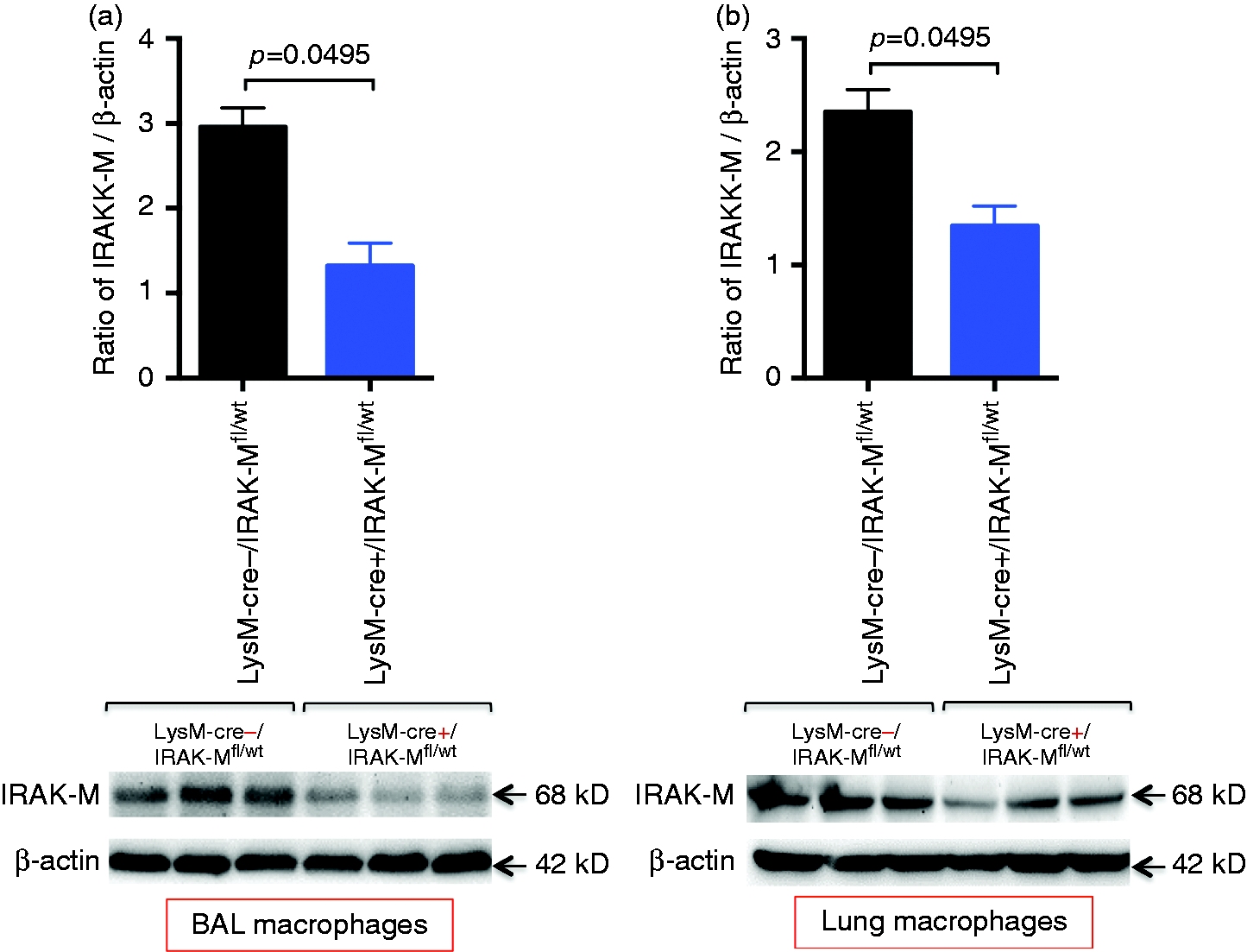
To confirm the cre genotyping data, cre mRNA expression was compared between cells isolated from LysM–Cre+/IRAK-Mfl/wt and LysM–Cre–/IRAK-Mfl/wt mice. Cre expression was only seen in pAM and blood leukocytes from LysM–Cre+/IRAK-Mfl/wt mice (Figure 3a). However, cre was absent in tracheal epithelial cells from LysM–Cre+/IRAK-Mfl/wt mice, indicating that cre expression is restricted to cells of myeloid cell lineage. Importantly, IRAK-M mRNA expression followed the pattern of cre expression. In pAMs, the levels of IRAK-M mRNA (Figure 3b) and protein (Figure 4a) were markedly decreased in LysM–Cre+/IRAK-Mfl/wt mice compared with control mice. Moreover, IRAK-M protein was reduced in isolated lung macrophages of LysM–Cre+/IRAK-Mfl/wt mice (Figure 4b). IRAK-M mRNA trended to be lower in blood leukocytes of LysM–Cre+/IRAK-Mfl/wt mice. In contrast, IRAK-M mRNA levels in tracheal epithelial cells were similar between the two groups of mice (Figure 3b).
Detection of Cre and IRAM-M mRNA expression in LysM–Cre+/IRAK-Mfl/wt and LysM–Cre–/IRAK-Mfl/wt mice. Quantitative RT-PCR was performed to measure specifically mRNA of (a) Cre and (b) IRAK-M exon 3 in naïve pAM, blood leukocytes (blood leuk) and tracheal epithelial cells (TEC) from LysM–Cre+/IRAK-Mfl/wt and LysM–Cre–/IRAK-Mfl/wt mice. Data are expressed as means ± SEM (n = 3 mice/group). Reduced IRAK-M protein levels in macrophages of LysM–Cre+/IRAK-Mfl/wt mice. Macrophages from BAL and lung tissues were isolated from naïve LysM–Cre+/IRAK-Mfl/wt and LysM–Cre–/IRAK-Mfl/wt mice and processed for IRAK-M Western blot. Densitometric analysis of IRAK-M protein normalized by β-actin and Western blot images of IRAK-M protein from (a) BAL macrophages and (b) lung tissue macrophages. Data are expressed as means ± SEM (n = 3 mice/group).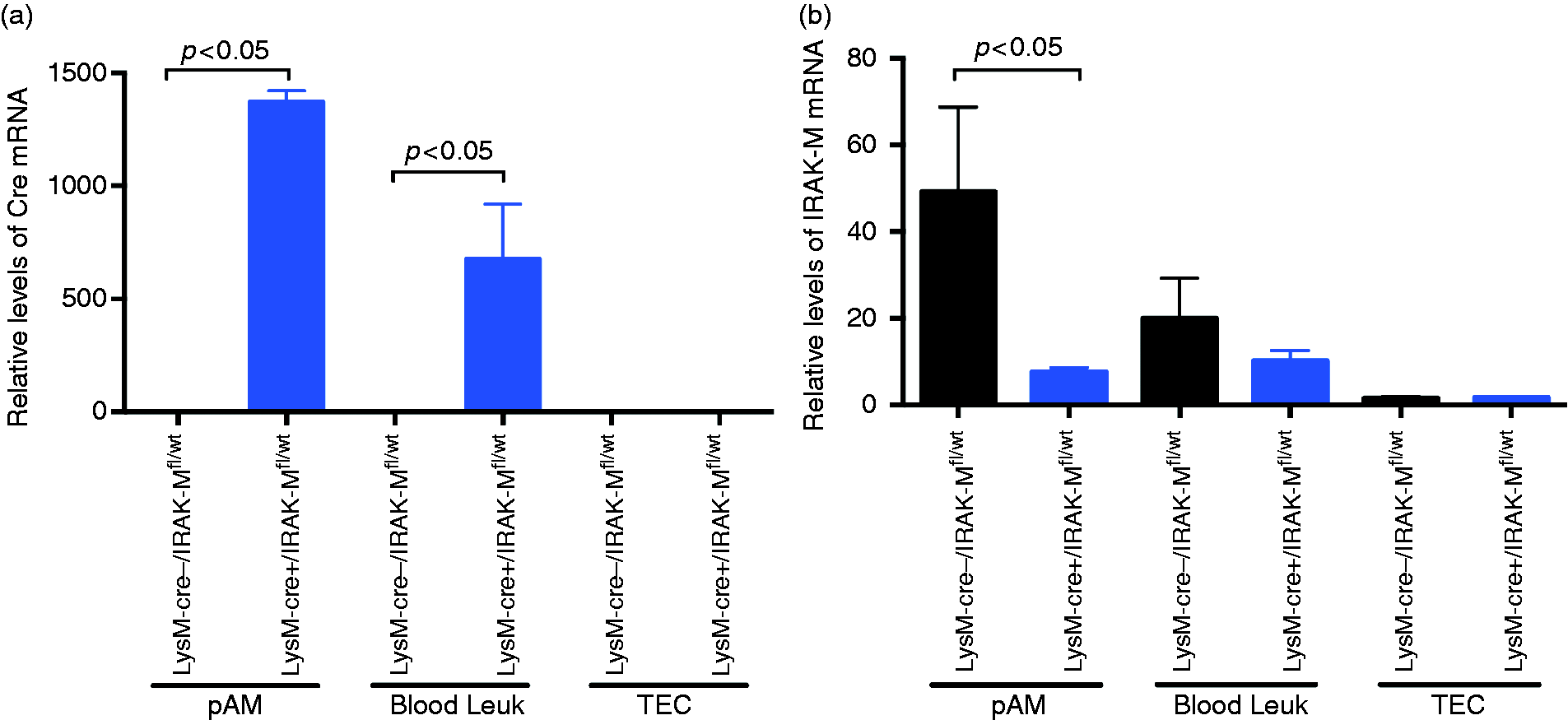
LysM–cre+/IRAK-Mfl/wt mice display exaggerated lung pro-inflammatory response to PA infection
Having shown deficient IRAK-M expression in myeloid cells of LysM–Cre+/IRAK-Mfl/wt mice, we determined if these mice respond to PA infection differently from the control LysM–Cre+/IRAK-Mfl/wt mice. As shown in Figure 5(a, b), in PBS-treated mice, IRAK-M deficiency in myeloid cells did not alter the number of leukocytes, including neutrophils, in BAL fluid. In contrast, with PA infection, total leukocytes, including neutrophils, were significantly increased in BAL fluid of LysM–Cre+/IRAK-Mfl/wt mice compared with the control mice. Neutrophils are the predominant cells following PA infection. The levels of KC, a chemoattractant for neutrophils, were consistent with the neutrophil data in BAL fluid (Figure 5c). BAL macrophages and lymphocytes were similar between the two groups of mice.
Increased lung inflammatory response in LysM–Cre+/IRAK-Mfl/wt mice with PA infection. (a) Leukocytes, (b) neutrophils and (c) KC were examined in BAL fluid of LysM–Cre+/IRAK-Mfl/wt and LysM–Cre–/IRAK-Mfl/wt mice after 1 d of intranasal PBS treatment or PA infection. The horizontal line indicates the median (n = 8–9 mice/group). (d) BAL macrophages from naïve LysM–Cre+/IRAK-Mfl/wt and LysM–Cre–/IRAK-Mfl/wt mice were isolated and stimulated with LPS (100 ng/ml) for 24 h. Supernatants were collected to measure TNF-α protein levels by ELISA. Data are expressed as means ± SEM (n = 5 mice/group).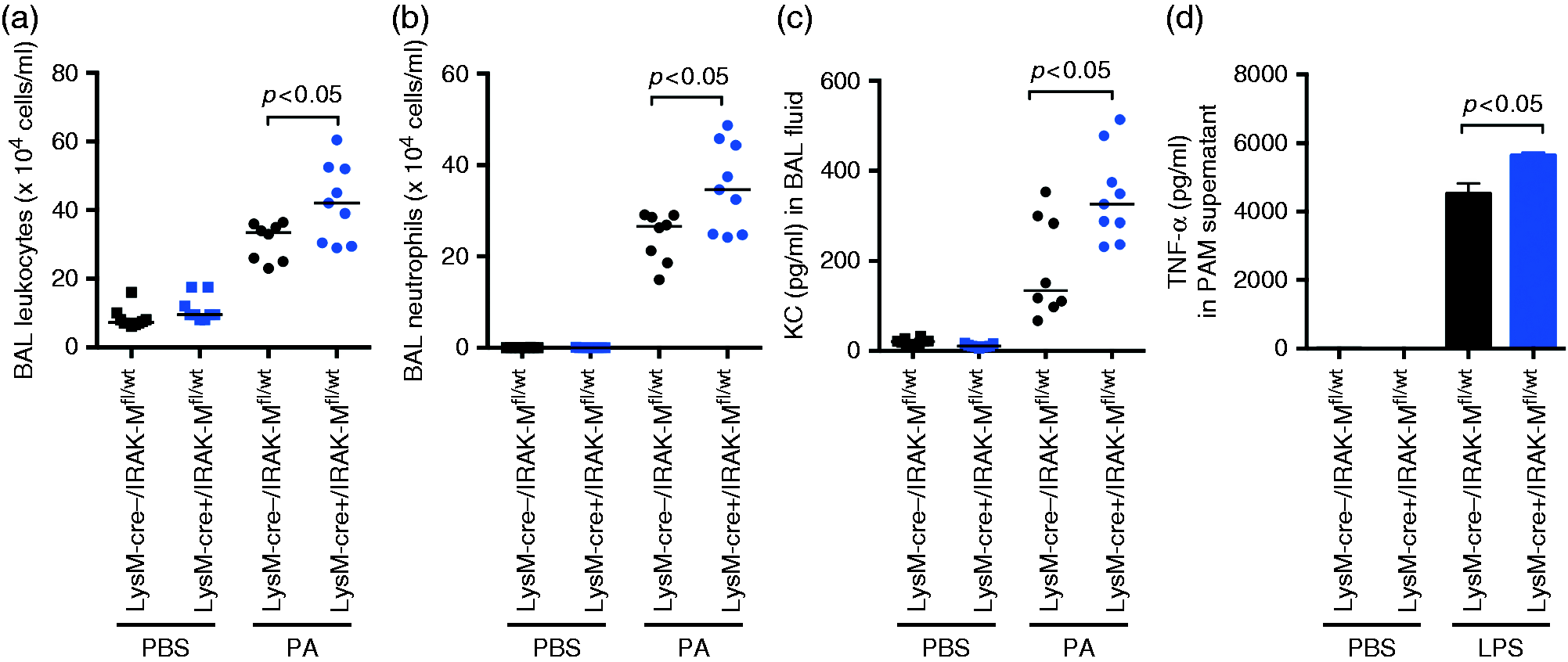
In line with BAL fluid inflammation data, LysM–Cre+/IRAK-Mfl/wt mice demonstrated greater lung tissue inflammation in response to PA infection, as represented by a higher histopathology score, than the control mice (Figure 6a). A shown in Figure 6b, LysM–Cre+/IRAK-Mfl/wt mice exhibited greater peribronchial/perivascular leukocyte infiltration and pneumonia.
Lung histopathology. Increased lung tissue inflammation (mainly neutrophils) in LysM–Cre+/IRAK-Mfl/wt mice with PA infection. Lung histopathology was evaluated in LysM–Cre–/IRAK-Mfl/wt and LysM–Cre+/IRAK-Mfl/wt mice after 1 d of intranasal PBS treatment or PA infection. (a) Histopathology scores in indicated mouse groups. (b) Photomicrographs of lung tissue sections stained with hemotoxylin and eosin. (a, b) PBS treatment in LysM–Cre–/IRAK-Mfl/wt and LysM–Cre+/IRAK-Mfl/wt mice, respectively. ((c, d) PA infection in LysM–Cre–/IRAK-Mfl/wt and LysM–Cre+/IRAK-Mfl/wt mice, respectively. Original magnification, ×200.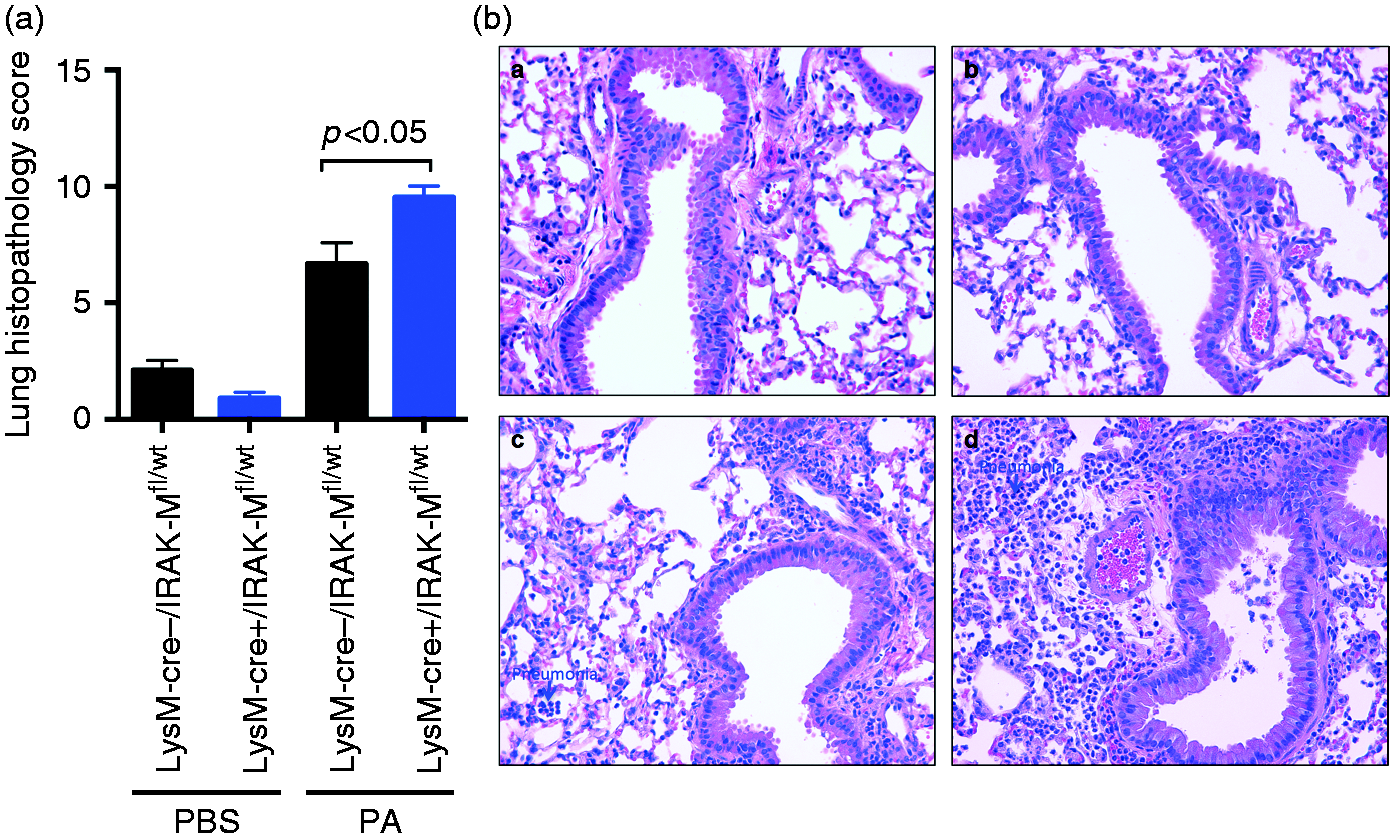
We further confirmed that isolated alveolar macrophages from LysM–Cre+/IRAK-Mfl/wt mice (no PA infection) had increased production of pro-inflammatory cytokine TNF-α in response to ex vivo LPS compared with control mice (Figure 5d).
LysM–Cre+/IRAK-Mfl/wt mice demonstrated reduced PA load in the lung
To determine the effects of myeloid cell IRAK-M deletion on lung bacterial clearance, we measured PA load after 24 h of bacterial infection. LysM–Cre+/IRAK-Mfl/wt mice demonstrated significantly lower levels of PA in the lung (Figure 7a) and BAL fluid (Figure 7b) compared with the control mice.
Decreased lung PA load in LysM–Cre+/IRAK-Mfl/wt mice. LysM–Cre+/IRAK-Mfl/wt and LysM–Cre–/IRAK-Mfl/wt mice were intranasally infected with PA for 24 h. PA in (a) lung tissue and (b) BAL fluid was cultured on agar plates and counted. The horizontal line indicates the median (n = 8–9 mice/group).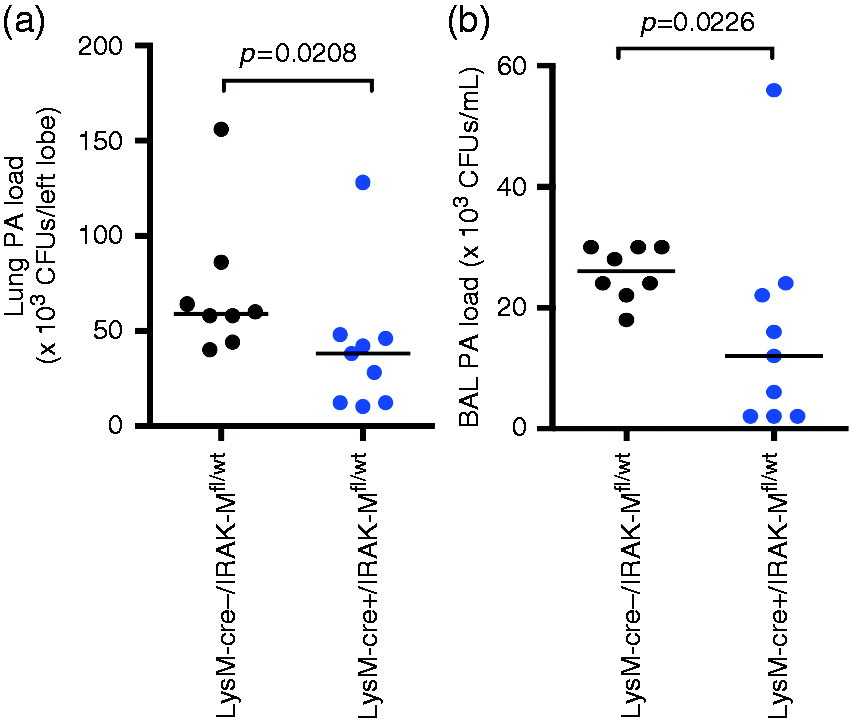
Mechanisms of reduced lung PA load in LysM–Cre+/IRAK-Mfl/wt mice
Neutrophils are critical to the clearance of bacteria from the lungs using multiple mechanisms, including the production and release of MPO.
23
To confirm if increased neutrophils in PA-infected LysM–Cre+/IRAK-Mfl/wt mice were associated with increased release/activity of MPO, we measured MPO in BAL fluid. Compared with the low level of MPO in PBS-treated mice, MPO activity was increased following PA infection. Importantly, LysM–Cre+/IRAK-Mfl/wt mice, compared with the control mice, had higher levels of MPO in BAL fluid upon PA infection (Figure 8).
Increased MPO activity in BAL fluid of LysM–Cre+/IRAK-Mfl/wt mice with PA infection. LysM–Cre+/IRAK-Mfl/wt and LysM–Cre–/IRAK-Mfl/wt mice were intranasally infected with PA for 24 h. BAL fluid was collected for quantifying MPO activity. The horizontal line indicates the median (n = 8–9 mice/group).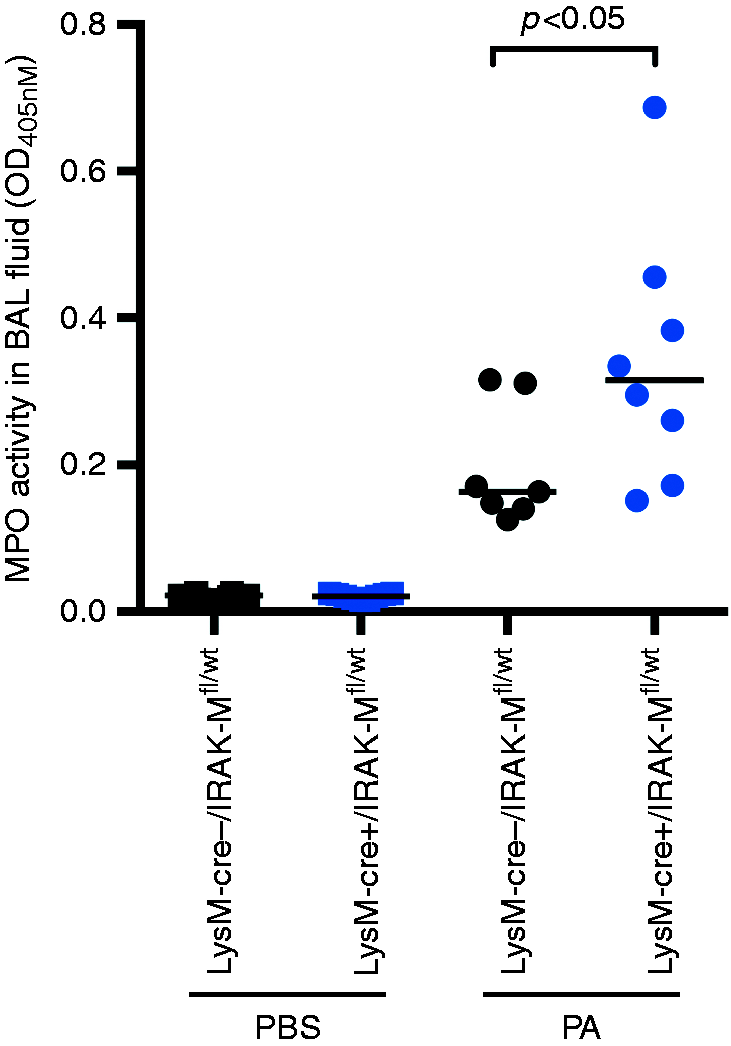
To determine if increased MPO activity in LysM–Cre+/IRAK-Mfl/wt mice is, in part, responsible for reduced PA load, we used filtered mouse BAL fluid with or without a selective MPO inhibitor ABAH to test the antimicrobial effect of BAL fluid against PA. No bacterial growth was found in filtered BAL fluid. Therefore, the culture experiment using exogenous PA indicates the antibacterial effect of BAL fluid. As shown in Figure 9a, in the absence of ABAH (DMSO control), BAL fluid from LysM–Cre+/IRAK-Mfl/wt mice significantly reduced PA growth compared with the control mice. In the presence of ABAH (30 min pre-treatment), such reduction of PA load in BAL fluid of LysM–Cre+/IRAK-Mfl/wt mice disappeared. Notably, ABAH treatment of BAL fluid of both strains of mice increased PA levels. The specificity of ABAH was confirmed by reduced activity of recombinant mouse MPO (Figure 9b) and increased PA levels in the PA culture experiment (Figure 9c).
MPO-dependent antibacterial activity in BAL fluid of PA-infected mice. (a) BAL fluid from PA-infected (1 d) LysM–Cre+/IRAK-Mfl/wt and LysM–Cre–/IRAK-Mfl/wt mice was filtered to remove PA and then treated with DMSO (control) or with a selective MPO inhibitor, ABAH, for 10 min, followed by culture with exogenous PA (103 CFUs/ml) for 2 h. PA was plated on agar plates and counted. (b) Recombinant mouse MPO (rmMPO) was incubated with DMSO (control) or with a selective MPO inhibitor, ABAH, for indicated times. MPO activity was measured on a spectrometer and expressed as OD values. (c) rmMPO was treated with DMSO (control) or ABAH for 10 min, followed by culture with exogenous PA (103 CFUs/ml) for 2 h. PA was plated on agar plates and counted.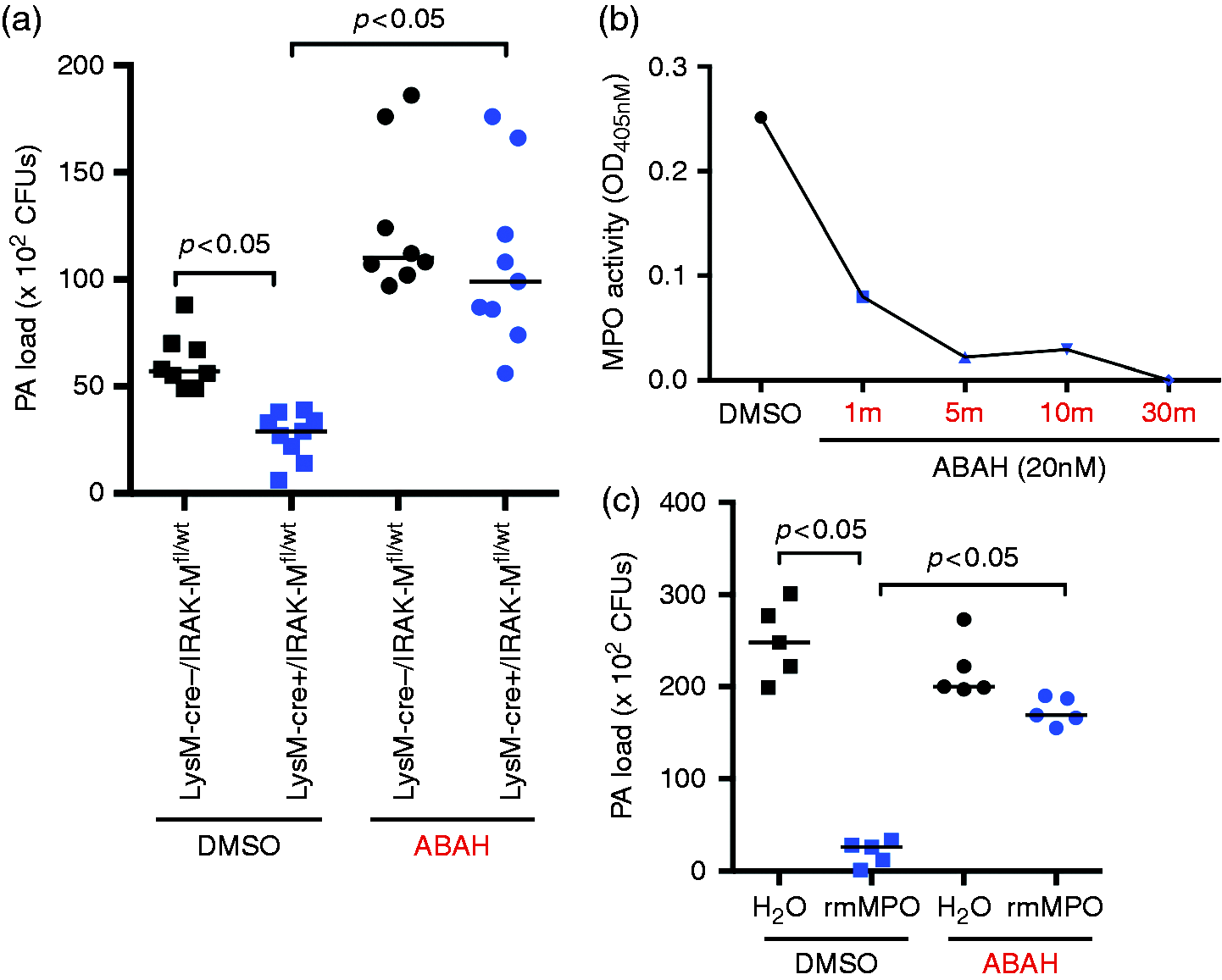
Discussion
To the best of our knowledge, this is the first study to generate mice with IRAK-M deficiency in myeloid cells using the cre–loxP system and validate them in a lung bacterial infection model.
Several key findings from the current study support the specificity of IRAK-M deletion in myeloid cells. Significantly reduced IRAK-M mRNA and/or protein expression were observed in isolated BAL and lung macrophages. The unchanged IRAK-M expression in non-myeloid cells such as isolated tracheal epithelial cells from LysM–Cre+/IRAK-Mfl/wt mice vs. the control mice further validated the cell type specificity of our conditional IRAK-M knockout mouse model using the cre–loxP system.
By using the intranasal PA infection model, we were able to demonstrate increased lung neutrophilic inflammation and decreased PA load in LysM–Cre+/IRAK-Mfl/wt mice. Our data are supported by a previous study in which whole-body IRAK-M knockout mice infected with intratracheal PA infection demonstrated greater pro-inflammatory response to PA infection, and less bacterial load than wild type mice. 12 As IRAK-M is also expressed in epithelial cells,24,25 a mouse model of myeloid cell-specific (e.g. macrophage) deletion of IRAK-M will be invaluable to dissect the contribution of myeloid cells vs. structural cells (e.g. epithelial cells) to host responses to pathogens that are detrimental to the health of patients.
A novel finding from our study is that deletion of IRAK-M in myeloid cells reduces lung PA load, in part, via enhanced MPO production. IRAK-M up-regulation has been documented in macrophages of patients with sepsis and other conditions,12,26 and in airway epithelial cells of patients with asthma. 18 It is possible that increased IRAK-M expression under diseased conditions would compromise host defense functions. Different pathogens vary in their tissue tropism of infection. Therefore, understanding the role of IRAK-M in host defense mechanisms by different cell types would greatly improve our approach to more effectively and precisely eliminate the detrimental effects of pathogen infections.
One intriguing finding is that IRAK-M mRNA level in blood leukocytes of LysM–Cre+/IRAK-Mfl/wt mice is not significantly lower than that of the control mice (Figure 3b). We speculate that lymphocytes in the mixed blood leukocyte preparation might have reduced the difference of IRAK-M mRNA expression in myeloid cells between the two strains of mice. We also noticed the degree of cre-mediated IRAK-M mRNA reduction in lung macrophages was not the same as protein reduction. This may be explained by the fact that mRNA measurement by quantitative PCR is more sensitive than protein measurement by Western blot. Additionally, previous a study suggests that various factors, such as transcriptional splicing, post-transcriptional splicing, translational modifications, translational regulation and protein complex formation, also contribute to varying degree of correlations between mRNA and protein expression. 27
There are several limitations to our study. First, we did not examine each type of myeloid cells (e.g. neutrophils) and non-myeloid cells (e.g. lymphocytes) to further test the cellular specificity of cre-mediated deletion of IRAK-M expression. Nonetheless, given the specificity and the most widely used nature of the LysM promoter to drive cre overexpression and subsequent gene deletion in myeloid cells, especially macrophages,28,29 our model offers a novel opportunity to define the contribution of IRAK-M from myeloid cells to host defense against pathogen infection, and perhaps other immunological processes. Second, our novel mouse model represents heterozygous IRAK-M deletion in myeloid cells. This may be owing to the fact that the IRAK-M and LysM genes are located in close proximity on chromosome 10. As a result, it is unlikely that a crossover event will take part between these two genes and achieve complete deletion of IRAK-M. This hypothesis was confirmed by our repeated efforts in breeding these mice but with the same genotyping result in that only heterozygous IRAK-M deleted mice (LysM–Cre+/IRAK-Mfl/wt) were generated. Other approaches such as the use of CSF1R promoter will be considered to achieve, possibly, homozygous deletion of IRAK-M in myeloid cells. 28 Lastly, we only evaluated the functional consequence of IRAK-M deletion in LPS-stimulated alveolar macrophages. Although alveolar macrophages are invaluable for evaluating the function of myeloid cell IRAK-M, future studies are warranted to determine the function of IRAK-M in other types of myeloid cells, such as neutrophils in the lung and blood.
Our success in generating myeloid cell-specific deletion of IRAK-M will enable us to extend the PA infection model to other pathogen infection models. As other species of bacteria (e.g. Haemophilus influenzae) or viruses (e.g. human rhinovirus) are associated with lung diseases,30–33 it would be necessary to test whether IRAK-M in myeloid cells exerts immunomodulatory functions against those respiratory pathogens. The current study is focused on the use of LysM–cre mice, but crossing IRAK-M floxed mice with those expressing a specific gene promoter in other cell types will broaden our understanding of IRAK-M function. For example, crossing IRAK-M floxed mice with CD11c–cre mice will allow us to study the functional consequence of IRAK-M deletion in both myeloid cells and dendritic cells, or with Tie2–cre mice to study IRAK-M function in all types of leukocytes.
In summary, our current study has significantly advanced our understanding of the in vivo function of myeloid cell IRAK-M during lung bacterial infection. Our novel IRAK-M conditional knockout mouse model will allow investigators to look into the role of IRAK-M in any cell types of interest, such as lung epithelial cells.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the grants from the National Institutes of Health, R01 HL122321, R01 AI106287, R01 HL125128 and U19AI125357.