
Abstract
Inflammatory signaling pathways induced by Helicobacter pylori remain unclear, having been studied mostly on cell-line models derived from gastric adenocarcinoma with potentially altered signaling pathways and nonfunctional receptors. Here, H. pylori-induced signaling pathways were investigated in primary human gastric epithelial cells. Inflammatory response was analyzed on chemokine mRNA expression and production after infection of gastric epithelial cells by H. pylori strains, B128 and B128ΔcagM, a cag type IV secretion system defective strain. Signaling pathway involvement was investigated using inhibitors of epidermal growth factor receptor (EGFR), MAPK, JAK and blocking Abs against TLR2 and TLR4. Inhibitors of EGFR, MAPK and JAK significantly reduced the chemokine mRNA expression and production induced by both H. pylori strains at 3 h and 24 h post-infection. JNK inhibitor reduced chemokine production at 24 h post-infection. Blocking Abs against TLR2 but not TLR4 showed significant reduction of chemokine secretion. Using primary culture of human gastric epithelial cells, our data suggest that H. pylori can be recognized by TLR2, leading to chemokine induction, and that EGFR, MAPK and the JAK/STAT signaling pathways play a key role in the H. pylori-induced CXCL1, CXCL5 and CXCL8 response in a cag pathogenicity island-independent manner.
Introduction
Helicobacter pylori infects half of the human population, and leads to local gastric mucosal inflammation. 1 Chronic H. pylori infection can cause gastric ulcers and non-cardia gastric cancer. 2 Pro-inflammatory signaling pathways induced by H. pylori have been studied in many reports, but remain unclear and at times contradictory.3–6 Multiple bacterial virulence factors and cellular molecules seem to be involved in this signaling. 4 In fact, the type IV secretion system (T4SS) may cause a pro-inflammatory response either by injected virulence factors such as cagA7,8 or peptidoglycan,9,10 or by direct contact with target cells itself. 11 It has been reported that peptidoglycan recognized by the intracellular receptor NOD1 or a bacterial enzyme, urease, could result in NF-κB activation and CXCL8 (also known as IL-8) production.9,12 The outer membrane protein, OipA, is involved in activation of signal inducer and activator of transcription A (STAT1), leading to CXCL8 secretion. 13 In addition, epidermal growth factor receptor (EGFR) pathway seems to play an important role in CXCL8 induction by H. pylori. 14 Keates et al. 15 have demonstrated that CXCL8 production induced by H. pylori involves transactivation of the EGF receptor via an endogenous ligand, the heparin-binding epidermal growth factor (HB-EGF), released by a matrix metalloprotease. MAPK and JAK/STAT pathways have also been implicated in the inflammatory response induced by H. pylori.16–20 Thus, different signaling pathways seem to be involved in CXCL8 production after gastric epithelial cell infection by H. pylori. Moreover, implication of TLRs, especially TLR2 and TLR4, in the recognition of H. pylori-associated molecular patterns is controversial.5,6 H. pylori has a LPS with low bioactivity,6,21,22 which could be recognized by TLR2 rather than TLR4 for some authors,21,23,24 or by neither TLR4 nor TLR2 for others. 22
Nevertheless, most of these reports have come from analyses performed on cell-line models derived from gastric adenocarcinoma. These cell lines were found to have lost some of the key elements in the signaling pathways potentially involved in cellular responses to H. pylori infection. For example, the AGS cell line lacks TLR2 and the myeloid differentiation factor 2 (MD2) cofactor, which is essential for LPS recognition by TLR4.23,24 To obtain a more physiological model, a primary culture of human gastric epithelial cells has been developed from sleeve-gastrectomy surgical specimens. 25 The aim of this study was to investigate the involvement of major signaling pathways in the CXCL1, CXCL5 and CXCL8 response of primary human gastric epithelial cells (PGEC) induced by H. pylori by considering the role of the cag Pathogenicity Island (cagPAI) bacterial virulence factor. It was shown that PGEC are more reactive to H. pylori infection than AGS cells, secreting a wider panel of pro-inflammatory chemokines, such as CXCL1 and CXCL5. 26 The previous study showed that induction of inflammatory mediators was dependent on H. pylori cagPAI substrate at an early stage of infection. However, this induction of inflammatory chemokines appeared independent of H. pylori cagPAI substrate at 24 h post-infection, whereas other studies on AGS cells or epithelial progenitor cells, supported the hypothesis that CXCL8 production is highly dependent on a functional cagPAI.9,26–29 In this study, using inhibitors of EGFR, MAPK and JAK/STAT signaling pathways and blocking Abs against TLR2 and TLR4, the involvement of the signaling pathways in the H. pylori-induced chemokine response was studied in both PGEC and AGS cellular models.
Materials and methods
Primary culture of human gastric epithelial cells
The protocol for culture was optimized from our previously described methods. 25 The use of stomach samples for this study was approved by the ethics committee of Poitiers hospital (Poitiers, France). Under fully informed consent, human stomachs of H. pylori-negative subjects were collected from surgical specimens of sleeve-gastrectomy. After washes, the mucosa was dissected from underlying submucosa and minced into small fragments. Then, after enzyme digestion with 0.5 mg/ml collagenase B (Roche, Basel, Switzerland), 2.5 mg/ml pronase (Roche) and 3 U/ml dispase (Sigma-Aldrich, St. Louis, MO, USA) for 30 min at 37℃ and digestion in trypsin-0.05% EDTA (Gibco, Waltham, MA, USA) for 15 min at 37℃, the cell suspension collected was filtered through a 250-µm Nitex mesh. Cells were washed twice, centrifuged at 400 g for 5 min and filtered again through a 100-µm Nitex mesh. Primary human gastric cells were seeded at a density of 5 × 105–1 × 106 cells/well in 24-well collagen I-coated culture plates (Becton Dickinson, Franklin Lakes, NJ, USA). The culture medium optimized for the adhesion of viable cells consisted of CnT-Basal Medium 1 (CELLnTEC, Bern, Switzerland) supplemented with 10% FCS (Sigma-Aldrich), 5 ng/ml EGF (Gibco), 15 ng/ml hepatocyte growth factor (HGF), 50 ng/ml insulin growth factor, 5 ng/ml fibroblast growth factor (FGF)2, 1 ng/ml FGF7 (all Miltenyi Biotec, Bergisch Gladbach, Germany), 10 nM gastrin (Sigma-Aldrich), 10 µg/ml insulin (Sigma-Aldrich), 10 µg/ml TGF-β1 (Peprotech, Rocky Hill, NJ, USA), 50 U/ml penicillin and 50 µg/ml streptomycin (Gibco). After 24 h incubation, culture medium was replaced by Ham’s F12/DMEM (v/v) medium (Gibco) supplemented with 10% FCS, 5 ng/ml EGF, 15 ng/ml HGF, 5 ng/ml FGF2, 1 ng/ml FGF7, 50 U/ml penicillin and 50 µg/ml streptomycin. Culture medium was exchanged every 2 d and the cultures were incubated at 37℃ in a humidified atmosphere with 5% CO2. PGEC were cultured to 80% of confluence before infection assays with H. pylori.
Human gastric cell line culture
AGS cell line (ATCC® number: CRL 1739™) was cultured in 75 cm2 flasks in DMEM supplemented with 10% FCS, 50 U/ml penicillin and 50 µg/ml streptomycin. AGS cells were seeded at a density of 4 × 105 cells/well in 24-well culture plates. The cultures were maintained at 37℃ in a humidified atmosphere with 5% CO2 until 80% confluence was obtained.
Bacterial culture
The H. pylori strains used throughout the study were B128 and B128ΔcagM strains. H. pylori B128 strain (cagA, vacA: s1/m2) was isolated from a patient with gastric ulcer disease.30,31 This strain contains an entirely functional cagPAI. B128ΔcagM, an isogenic mutant, was obtained by natural transformation, allelic exchanges and insertion of a chloramphenicol resistance cassette. 9 PCR of the cagM gene empty site was performed to confirm the mutagenesis. Deletion of the cagM gene renders H. pylori T4SS dysfunctional in such a way that B128ΔcagM strains lose their ability to perform efficient translocation of CagA or peptidoglycan into epithelial cells. 9 H. pylori strains were routinely cultured on Columbia blood agar supplemented with antibiotics (Skirrow supplement; Oxoid, Altrincham, UK) and incubated for 48 h at 37℃ in microaerobic conditions using CampyGen bags (Oxoid). Before cell-infection assays, bacteria were cultured on blood sheep agar medium without antibiotics for 24 h. Then, suspensions of H. pylori B128 and B128ΔcagM strains were prepared in the cell culture medium. Bacteria were added to cells at a MOI equal to 100 bacteria per cell. Bacterial concentrations were determined by measuring the OD of the culture at 600 nm. In addition, CFU counts of H. pylori were performed.
Cell infection assays with H. pylori strains
PGEC and AGS cells were incubated in Ham’s F12/DMEM culture medium without growth factors, FCS or antibiotics for 12 h prior to infection. Cell monolayers were then infected with H. pylori B128 or B128ΔcagM strains for 3 h or 24 h at 37℃ in a humidified atmosphere of 5% CO2. Culture supernatants were collected, centrifuged (400 g, 5 min, 20℃) and stored at –80℃ until used. Cultures without bacteria were used as controls. To assess the involvement of the different signaling pathways, PGEC cells were pre-treated for 2 h by different inhibitors and Abs before being infected by H. pylori. The working concentrations of inhibitors were selected taking into consideration those recommended by the manufacturer, the data from numerous previous studies using the same inhibitors on gastric cell lines,10, 11,15,19,32,33 and the results of cytotoxicity assays on gastric cells. The role of EGFR signaling in inflammatory response induced by H. pylori was investigated with tyrphostin AG1478 (2 µmol/l; Sigma-Aldrich), a specific inhibitor of EGFR tyrosine kinase, and gefitinib (10 µmol/l; Invivogen, San Diego, CA, USA), an inhibitor of EGFR tyrosine kinase and receptor-interacting protein 2. Batimastat, a matrix metalloprotease inhibitor, was also used. To explore the involvement of the MAPK pathway, PGEC cells were incubated with an inhibitor of MEK-1/2, U0126 monoethanolate (10 µmol/l; Sigma-Aldrich) or an inhibitor of JNK, SP600125 (10 µmol/l; Sigma-Aldrich). A specific inhibitor of JAK, JAK inhibitor 1 (Calbiochem, Billerica, MA, USA), was used at 1 µmol/l. TLR signaling was investigated with neutralizing monoclonal Abs against human TLR2 and TLR4 (TLR2 IgA2 and TLR4 IgG1, 5 µg/ml; Invivogen) and corresponding istotype control Abs [human IgA2, 5 µg/ml (Invivogen) and mouse IgG1, 5 µg/ml (R&D systems)], as well as with a specific TLR2 ligand, lipoteichoic acid from Staphylococcus aureus, or a specific TLR4 ligand, synthetic monophosphoryl lipid A, from Escherichia coli (1 µg/ml; Invivogen). Phosphatidylinositol-3-kinases inhibitor AS6004850 (10 µmol/l; Sigma-Aldrich), PKC inhibitor Gö6983 (10 µmol/l; Sigma-Aldrich), IL-1 receptor-associated kinases 1/4 inhibitor I (2.5 µmol/l; Sigma-Aldrich) and NOD1 inhibitor Nodinitib 1 (30 µmol/l; Cayman Chemical, Ann Arbor, MI, USA) were also used in this study.
Cytotoxicity assay
Cytotoxicity of inhibitors was evaluated with a Cell proliferation kit I (MTT) (Roche) on AGS cells, as recommended by the manufacturer, in five independent assays. Absence of cytotoxicity of the inhibitors at the working concentrations was confirmed by Trypan Blue exclusion on PGEC after an incubation period of 24 h.
RNA extraction
Total RNA extraction from PGEC and AGS cells was performed using the Nucleo-Spin XS RNA extraction kit according to the manufacturer’s instructions (Macherey-Nagel). RNA was eluted in 10 µl of RNAse-free water supplemented with 40 units of RNaseout® (Invitrogen). RNA concentrations and purity were determined using the Nanodrop 2000 spectrophotometer (Thermo Scientific).
Reverse transcription and real-time PCR analysis
Total RNA (2 µg) was reverse transcribed using SuperScript II kit (Invitrogen) according to the manufacturer’s instructions. Quantitative RT-PCR was performed in 96-well plates using LightCycler-FastStart DNA MasterPlusSYBR GREEN I kit (Roche) on LightCycler 480 (Roche). The reaction mixture consisted of 1 × DNA Master Mix (Applied Biosystems, Foster City, CA, USA), 1 μM forward and reverse primers designed using Primer 3 software, and 12.5 ng cDNA template in a total volume of 10 µl. PCR conditions were as follows: 5 min at 95℃, 40 amplification cycles comprising 20 s at 95℃, 15 s at 64℃ and 20 s at 72℃. Samples were normalized with regard to two independent control housekeeping genes (GAPDH and B2M) and reported according to the ΔΔCT method as RNA fold increase: 2 -ΔΔCT = 2ΔCT stimulated- ΔCTunstimulated.
ELISA
Levels of CXCL1, CXCL5 and CXCL8 in cell culture supernatants were determined in duplicate using human ELISA development kits (R&D systems for CXCL1, and Peprotech for CXCL8 and CXCL5) in accordance with the manufacturers’ specifications.
Statistical analysis
The data presented constitute the average obtained from seven independently performed experiments with an SEM. Effects of the several inhibitors were analyzed comparing results with inhibitors and without inhibitors but with similar DMSO and bacterial concentrations. Statistical analysis of significance was calculated using Wilcoxon matched pairs test. P-Values ≤ 0.05 were considered as significant.
Results
Role of EGFR signaling pathway in PGEC CXCL8 response induced by H. pylori
The role of the EGFR pathway in chemokine expression of PGEC induced by H. pylori B128 and B128ΔcagM strains has been studied using two inhibitors of EGFR tyrosine kinase: gefitinib (10 µM) and AG1478 (2 µM). As previously reported using the PGEC model,
25
the B128ΔcagM strain with a nonfunctional T4SS induced a weaker chemokine expression than the wild type strain at an early stage of infection and AG1478 inhibited chemokine mRNA expression (Figure 1 and Supplementary Figure S1). On the whole, EGFR tyrosine kinase inhibitors significantly reduced CXCL8 mRNA expression induced by both H. pylori strains at 3 h post-infection (Figure 1a, b). In addition, chemokine production by PGEC was examined after 24 h of infection by H. pylori in presence of these inhibitors (Figure 1d, e). In agreement with our previous study,
25
the CXCL8 levels induced by both B128 and B128ΔcagM strains were similar at 24 h post-infection (Supplementary Figure S1). Gefitinib and AG1478 significantly decreased CXCL8 secretion induced by H. pylori. Similar results were obtained with AGS cells, as both EGFR inhibitors significantly reduced H. pylori induction of CXCL8 mRNA expression at 3 h post-infection and CXCL8 production after 24 h of infection (Supplementary Figure S2). EGFR pathway seemed to be activated in a cagPAI substrate-independent manner considering that inhibitors resulted in significant inhibition of chemokine expression and production induced by both H. pylori B128 and B128ΔcagM strains. In addition, EGFR expression was not significantly modulated by H. pylori after 24 h of infection (Supplementary Figure S3). To investigate the hypothesis that EGFR activation could result from the release of endogenous ligand from PGEC by metalloprotease, we used batimastat (1 µg/ml), a broad-spectrum metalloprotease inhibitor. A significant diminution of CXCL8 mRNA expression and secretion induced by both H. pylori strains was observed in presence of batimastat (Figures 1c, f).
CXCL8 mRNA expression and secretion after infection of gastric epithelial cells by H. pylori for 3 h and 24 h, respectively, in the presence or absence of specific inhibitors of EGFR pathway (gefitinib and AG1478) and metalloprotease (batimastat). Cells were infected by H. pylori B128 or B128ΔcagM at a MOI of 100 for 3 h or 24 h after 2 h incubation with or without inhibitor: (a, d) gefitinib 10 µM or (b, e) AG1478 2 µM or (c, f) batimastat 1 µg/ml. qRT-PCR analysis was carried out on total RNA of seven independent cultures from different patients. mRNA expression levels are expressed as the fold increase above unstimulated cultures. Protein concentrations (pg/ml) were measured in culture supernatants of seven independent cell cultures from different patients by ELISA assays. Data are represented as mean + SEM. *P < 0.05 compared with control without inhibitor.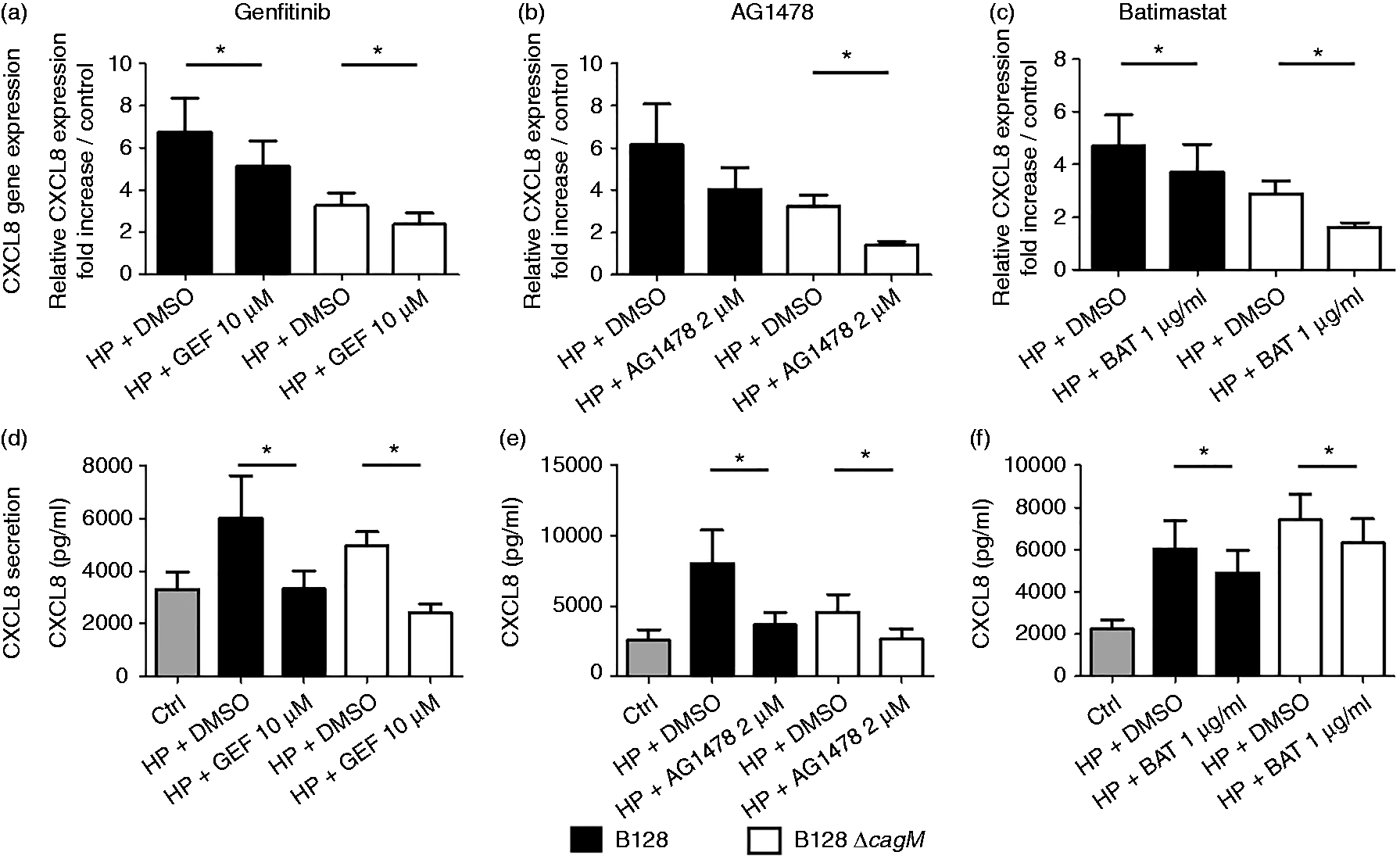
Involvement of the MAPK and the JAK/STAT signaling pathways
An ERK signaling pathway inhibitor, U0126 (10 µM), significantly reduced the CXCL8 mRNA expression induced by both H. pylori strains at 3 h post-infection (Figure 2a). However, the JNK signaling pathway inhibitor SP600125 at a final concentration of 10 µM did not reduce the CXCL8 mRNA expression induced by H. pylori strains (Figure 2b). Nonetheless, chemokine production by PGEC after 24 h of infection by H. pylori was significantly decreased in the presence of U0126 and SP600125 inhibitors (Figures 2d, e). Inhibition of the CXCL8 secretion induced by both strains of H. pylori was more pronounced in presence of U0126 than in presence of SP600125.
CXCL8 mRNA expression and secretion after infection of gastric epithelial cells by H. pylori for 3 h and 24 h, respectively, in the presence or absence of specific inhibitors of the MAPK and JAK pathways. Cells were infected by H. pylori B128 or B128ΔcagM at a MOI of 100 for 3 h or 24 h, after 2 h incubation with or without inhibitor: (a, d) U0126 10 µM (MEK1/2 inhibitor), (b, e) SP600125 10 µM (JNK inhibitor) or (c, f) JAK-1 inhibitor 1 µM. qRT-PCR analysis was carried out on total RNA of seven independent cultures from different patients. mRNA expression levels are expressed as the fold increase above unstimulated cultures. Protein concentrations (pg/ml) were measured in culture supernatants of seven independent cell cultures from different patients by ELISA assays. Data are represented as mean + SEM. *P < 0.05 compared with control without inhibitor.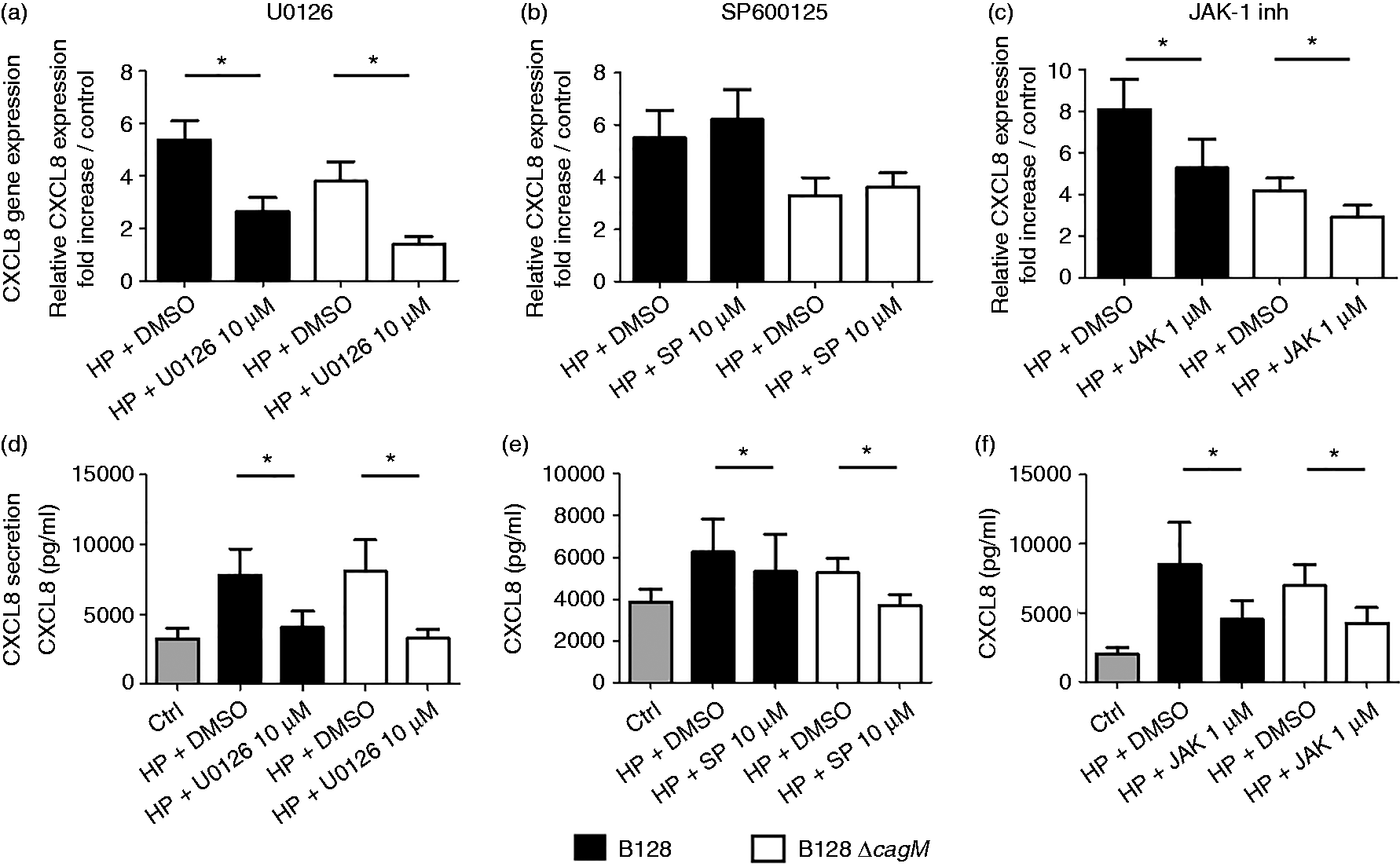
In addition, JAK1 inhibitor significantly reduced H. pylori-induced CXCL8 mRNA expression at 3 h post-infection (Figure 2c), as well as CXCL8 secretion (Figure 2f). ERK inhibitor, JNK inhibitor and JAK1 inhibitor have also been used on AGS cells, showing similar results on chemokine expression and production induced by both H. pylori strains (Supplementary Figure S2). Moreover, inhibition profiles of gene expression at 3 h post-infection or chemokine secretion at 24 h post-infection were relatively similar with both H. pylori strains B128 and B128ΔcagM, suggesting that activation of ERK, JNK and JAK pathways was not related to the cagPAI virulence factor. In addition, PI3K and NOD1 inhibitors were tested in the initial screening but did not show any real impact on H. pylori-induced chemokine expression, suggesting that these pathways play a minor role in PGEC inflammatory reactions at an early stage of infection (data not shown).
Role of TLR2 and TLR4 in PGEC CXCL8 response to H. pylori infection
The role of TLR2 and TLR4 receptors in inflammatory signaling pathways induced by H. pylori on PGEC was investigated using human TLR2 and TLR4 blocking Abs respectively. These Abs efficiently inhibited the PGEC chemokine production induced by corresponding specific TLR agonists (Supplementary Figure S4) and corresponding isotype control Abs have no impact on inflammatory response of either uninfected or infected cells (Supplementary Figure S5). On the whole, anti-TLR2 Ab significantly reduced the CXCL8 mRNA expression induced by both H. pylori strains at 3 h post-infection (Figure 3a). In addition, the CXCL8 secretion levels induced by H. pylori were significantly reduced in presence of anti-TLR2 Ab (Figure 3b). However, although anti-TLR4 blocking Ab slightly but significantly reduced CXCL8 mRNA expression induced by H. pylori B128ΔcagM, no inhibition of CXCL8 secretion by PGEC was observed in presence of this Ab after 24 h of infection by both H. pylori strains (Figure 3).
CXCL8 (a) mRNA expression and (b) secretion after infection of gastric epithelial cells by H. pylori for 3 h and 24 h, respectively, in the presence or absence of anti-TLR2 and anti-TLR4 blocking Abs. Cells were infected by H. pylori B128 or B128ΔcagM at a MOI of 100 for 3 h or 24 h, after 2 h incubation with or without blocking Abs: anti-TLR2 IgA2 5 µg/ml or anti-TLR4 IgG1 5 µg/ml. qRT-PCR analysis was carried out on total RNA of seven independent cultures from different patients. mRNA expression levels are expressed as the fold increase above unstimulated cultures. Protein concentrations (pg/ml) were measured in culture supernatants of seven independent cell cultures from different patients by ELISA assays. Data are represented as mean + SEM. *P < 0.05 compared with control without inhibitor.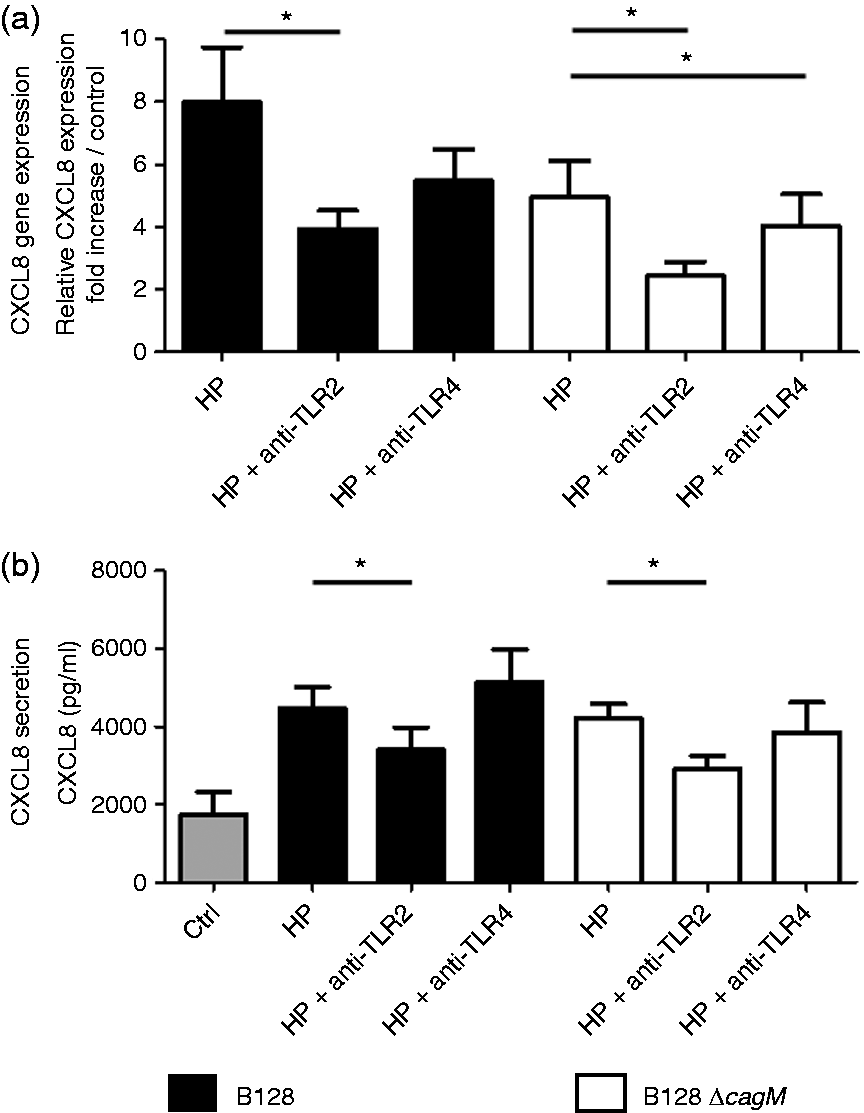
Involvement of the EGFR, MAPK, JAK/STAT and TLR2 signaling pathways in PGEC CXCL1 and CXCL5 response induced by H. pylori
As we previously reported that PGEC secreted high levels of CXCL1 and CXCL5 in response to H. pylori infection as compared with AGS model,
25
the involvement of the EGFR, MAPK, JAK/STAT and TLR2 signaling pathways in CXCL1 and CXL5 secretion induced by H. pylori was investigated. As observed for CXCL8 secretion, EGFR tyrosine kinase, ERK, JNK and JAK1 inhibitors, as well as anti-TLR2 Ab, decreased the CXCL1 and CXCL5 secretion induced by both H. pylori strains after 24 h (Figure 4a to j). None of the inhibitors at the working concentration used in this study has any impact on the cell viability or the basal chemokine level secreted by PGEC (data not shown).
CXCL1 and CXCL5 secretion after 24 h infection of gastric epithelial cells by H. pylori in the presence or absence of specific inhibitors of EGFR, MAPK and JAK pathways and anti-TLR2 blocking Abs. Cells were infected by H. pylori B128 or B128ΔcagM at a MOI of 100 for 24 h, after 2 h incubation with or without inhibitor: (a, f) gefitinib 10 µM, (b, g) U0126 10 µM (MEK1/2 inhibitor), (c, h) SP600125 10 µM (JNK inhibitor) or (d, i) JAK-1 inhibitor 1 µM; or blocking Abs: (e, j) anti-TLR2 IgA2 5 µg/ml. Protein concentrations (pg/ml) were measured in culture supernatants of seven independent cell cultures from different patients by ELISA assays. Data are represented as mean + SEM. *P < 0.05 compared with control without inhibitor.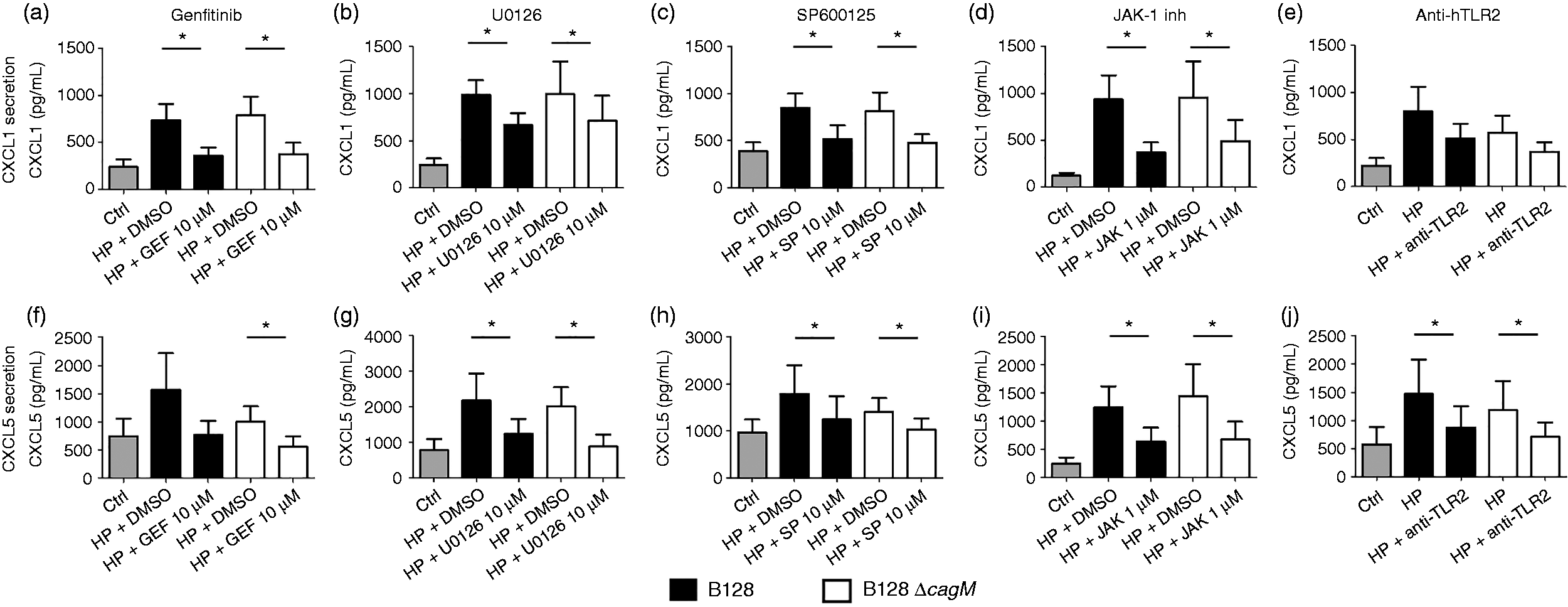
Discussion
Taken as a whole, our results suggest that inflammatory signaling pathways of primary gastric epithelial cells induced by H. pylori involve the EGFR, MAPK and JAK/STAT pathways. This study shows an early activation of these signaling pathways, except for the JNK pathway, which seems to play a subsequent role in chemokine induction in response to H. pylori infection. Moreover, all these pathways could lead to many forms of chemokine expression and secretion, including CXCL8, CXCL1 and CXCL5. Furthermore, TLR2 appeared to be involved in inflammatory reactions of PEGC to H. pylori infection.
CXCL8 was the main marker used to investigate the inflammatory response induced by H. pylori. Eftang et al. 34 reported that it was the most up-regulated gene of AGS cells after H. pylori stimulation. Previously, induction of several chemokines (CXCL1–CXCL3, CXCL5, CXCL8, CCL20) in PGEC infected by H. pylori has been observed. 25 In order to obtain an overview of the inflammatory signaling pathway induced by H pylori in PGEC, secretion of three chemokines that play a role in neutrophil recruitment and activation, CXCL1, CXCL5 and CXCL8, were studied. On the whole, our results suggest that similar signaling pathways are involved in the induction of the three chemokines. Inflammatory signaling pathways were investigated through primary culture of human gastric epithelial cells. This model is pertinent to study the H. pylori response of normal cells, especially with regard to TLR receptor involvement. Indeed, most of the studies have used different cell lines, derived from gastric carcinoma, which may have altered signaling pathways and nonfunctional receptors.7,10,15,17,18,21,24 For example, while expressing TLR4, AGS or MKN45 cell lines did not express the TLR4 co-receptor MD2, nor did they express TLR2.23,24 This may explain the controversial results on TLR implication in signaling pathways induced by H. pylori, obtained by transfection and overexpression of the analyzed TLRs on cancer cell lines.24,32,35,36 We report here the involvement of TLR2 but not of TLR4 in inflammatory response induced by H. pylori, a finding consistent with previous studies.23,24,37 However, the identity of the ligand able to activate TLR2 has yet to be determined. For some authors, LPS from H. pylori would be recognized by TLR2 rather than TLR4.21,24,38 More recently, Cullen et al., 22 using purified LPS on HEK cells transfected with different TLR machinery, suggested that H. pylori LPS does not activate TLR2, and conflicting results could come from contamination with lipoproteins or nucleic acids in LPS preparation. Moreover, H. pylori LPS recognition would tend to pass through TLR4, but was prevented by H. pylori lipid A 4’-phosphatase and lipid A 1-phosphatase. This phosphatase could be involved in H. pylori immune response evasion.
In the PGEC model, implication of EGFR, MAPK and JAK/STAT signaling pathways was consistent with other studies.13–15,17–20 The EGFR pathway has been shown to be an important signaling pathway after infection by H. pylori, which can lead to ERK signaling cascade,15,20 and also to JAK/STAT activation. 19 It has been proposed that transactivation of the EGFR can occur via shedding of an endogenous ligand, HB-EGF, through intervention of a metalloprotease. 15 Saha et al. reported that one of the T4SS pilus protein, CagL, mediates H. pylori-induced secretion of HB-EGF from AGS cells via activation of the metalloprotease ADAM17. 39 In this study, the metalloprotease inhibitor batimastat showed only a modest reduction of chemokine mRNA expression and protein production. This could suggest alternate ways for EGFR activation, or could also be due to incomplete inhibition of metalloprotease. Keates et al. 33 have pointed out that AGS infection by H. pylori also led to up-regulation of the expression of EGFR via transactivation of EGFR. In addition, Beswick and Reyes 14 have demonstrated that CXCL8 itself up-regulates EGFR expression and induce EGFR phosphorylation. Even though up-regulation of the EGFR was not observed in PGEC, this autocrine loop of amplification could be the cause of the uncontrolled inflammatory signal during chronic H. pylori infection.
With regard to the MAPK signaling pathway, the role of ERK and JNK kinases has been investigated. Whereas activation of ERK pathway seems to appear quickly, being observed mainly on CXCL8 mRNA expression at 3 h post-infection, activation of JNK pathway seems to occur later, as inhibition on chemokine mRNA expression was not seen after 3 h of infection but rather on chemokine secretion at 24 h post-infection. Several hypotheses have represented attempts to determine the pathways leading to ERK activation. For example, it was proposed that intracellular receptor NOD1, 10 protein CagA, 7 T4SS itself independently of NOD1 or CagA 11 and EGFR15,19,20 could be involved. The importance of the ERK pathway could come from its involvement in various signaling pathways, including the EGFR and TLR signaling pathways, as a pathway central to chemokine production. However, implication of the JAK/STAT pathway has been less widely investigated in inflammatory response. Bauer et al. 19 have shown that infection by H. pylori in AGS cells leads to human β-defensin 3 early induction via EGFR-dependent activation of MAPK and JAK/STAT signaling. Cha et al. 40 have also found that JAK1/STAT3 was activated after AGS cell infection by H. pylori, a phenomenon that may be involved in CXCL-8 expression via NF-κB activation. Signal transduction involved in inflammatory reactions induced by H. pylori resulted from a complex interplay of various bacterial factors, with a wide variety of host protein receptors.4,6 Primary gastric epithelial cell response to H. pylori revealed that multiple signaling pathways and receptors, including ERK, JNK, JAK/STAT, EGFR and TLR2, could be activated at an early post-infection stage. Cellular response seemed to be homogenous between individuals as the inhibitor’s effects were similar between PGEC obtained from different patients.
In our previous study on primary epithelial cells, 25 we found that the H. pylori B128ΔcagM strain, a T4SS-defective strain, elicits a weaker inflammatory chemokine mRNA induction than the wild type strain after 3 h of infection. But this difference was no longer observed at 24 h post-infection. This finding suggested that H. pylori cagPAI substrates were involved in eliciting an epithelial inflammatory response during the early phases of infection, but that after 24 h, inflammatory mediator production was largely due to cagPAI-substrate-independent virulence factors. In this work, no specific pathway induced only by H. pylori wild type B128 strain with a functional T4SS was clearly highlighted, suggesting that EGFR, MAPK and JAK/STAT pathways can be activated in a cagPAI-independent manner. Interestingly, it was demonstrated that induction of TLR2 expression by H. pylori switches cagPAI-dependent to cagPAI-independent signaling, leading to CXCL8 secretion. 36 Presence and involvement of functional TLR2 in PGEC response to H. pylori could explain the conflicting results obtained on AGS cells that do not express TLR2, where CXCL8 induction was primarily dependent on functional cagPAI. This study highlights the complexity of the inflammatory signaling pathways induced by H. pylori in primary cells and suggests that cagPAI substrates are not the main bacterial virulence factor responsible for chemokine secretion by PGEC.
Finally, our model of primary culture of human gastric epithelial cells has provided relevant complementary data on the signaling pathways involved in inflammatory response to H. pylori. This study supports the view that the bacterium can be recognized by TLR2 leading to chemokine induction and that EGFR, MAPK and JAK/STAT signaling pathways can play a key role in the H. pylori-induced inflammatory response. No difference in signaling pathway activation was found using strains with or without a functional cagPAI, thereby highlighting the complexity and multiplicity of the different signaling pathways that could lead to chemokine production during H. pylori infection.
Supplemental Material
Supplemental material for Inflammatory signaling pathways induced by Helicobacter pylori in primary human gastric epithelial cells
Supplemental Material for Inflammatory signaling pathways induced by Helicobacter pylori in primary human gastric epithelial cells by Cong Tri Tran, Magali Garcia, Martine Garnier, Christophe Burucoa and Charles Bodet in Innate Immunity
Footnotes
Acknowledgements
We thank Ivo G Boneca for providing the bacterial strains used in this study. We also thank Jean-Pierre Faure and Thierry Barthes for their kind collaboration that allowed us to obtain human gastric tissue. Finally, we thank Jeffrey Arsham, an American medical translator, for reviewing and editing our original English-language text.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.